
Graphene
Our calculations revealed that the roughness of graphene sheet could change the energy gain from epoxy group adsorption by a few tenths of electron volts. Such variations of energy are sufficient to switch between two scenarios as the two epoxy groups adsorption energy is close to the O2 molecule formation depending on local sheet curvature. Thus, the graphene sheet curvature can significantly affect its stability under UV/ozone treatment. Non-flat graphene is less stable to the ozone cleaning than the ideal one due to its convex regions are more reactive toward the epoxy group formation. We conclude that low roughness of epitaxial SiC graphene is a possible explanation of the enhanced robustness to ozone cleaning procedure compared to transferred graphene.
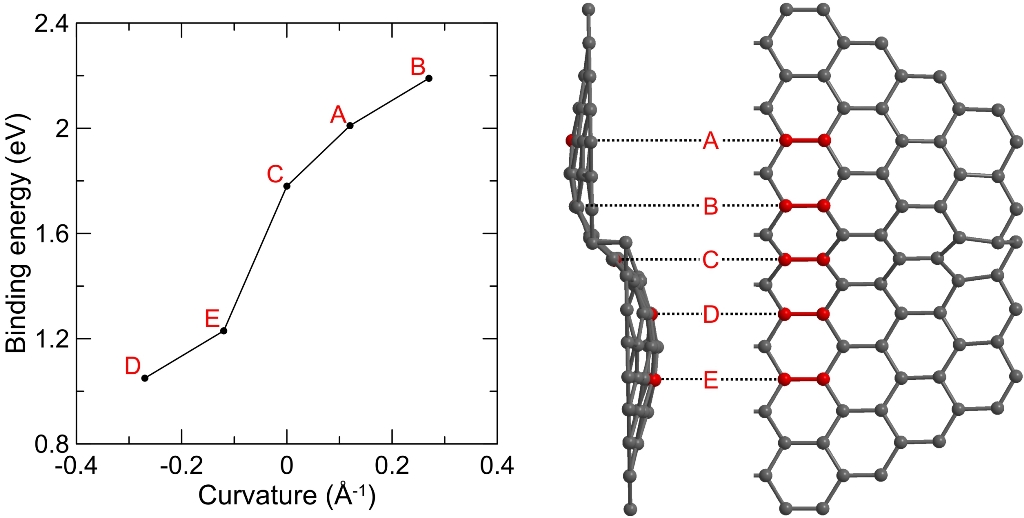
Figure 1. The curvature dependence of the binding energy of the epoxy group to graphene surface (left) and sine-shaped 44-rings graphene sheet atomic model (right): side view and plane half-view. Passivation by hydrogen atoms is not shown. Carbon atomic pairs for further epoxide formation are marked in red and labeled as A - E characters. Oxygen atom (not shown) is pointed toward a viewer at the plane half-view of the graphene fragment.
Using RO-B3LYP/6-311G(d) approach coupled with the nonorthogonal tight-binding model, we simulate the behavior of atomic hydrogens and hydroxyl groups on unstrained and stretched graphene sheets. The chemisorption energies obtained for H and OH radicals adsorbed on unstrained graphene are -0.69 eV and -0.27 eV, respectively. However, the further stretching of graphene leads to the decreasing of chemisorption energies. Surprisingly, chemisorption energies of H and OH radicals decrease in the similar manner: their decrease depending on the stretching is equal to 0.11 eV/%. Simultaneous chemisorption of two radicals in ortho or para positions leads to the energy gains of ~1 eV. The absolute values of these gains as well as energy differences between ortho and para configurations decrease as the graphene stretching rises.
Figure 2. Illustration of developed QM/QM model.
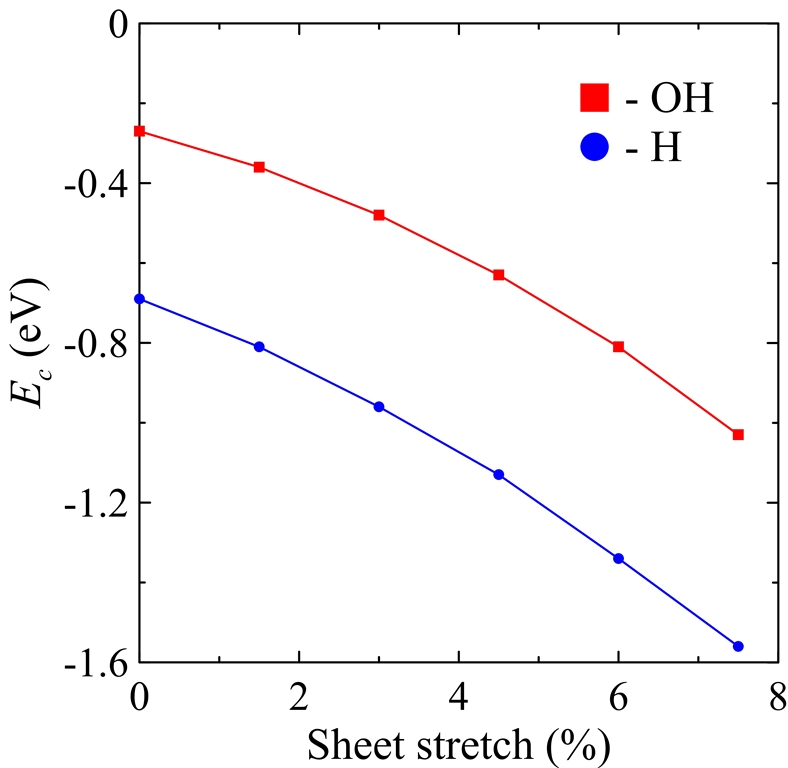
Figure 3. Chemisorption energies Ecs for single H and OH radicals (eV) on strained graphene as a function of sheet stretch e (%).
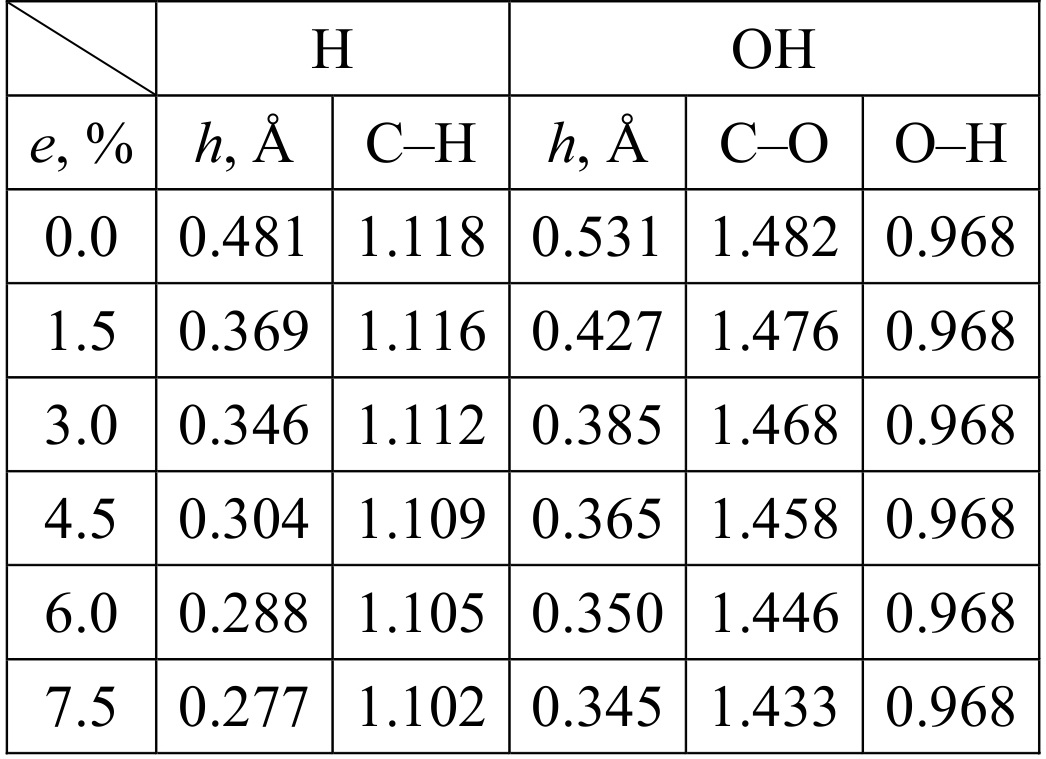
Table 1. Height of the doped carbon atom relative to the graphene plane h and the interatomic distances (in Å) for H and OH radicals chemisorbed on the stretched graphene as a function of stretch e (%).
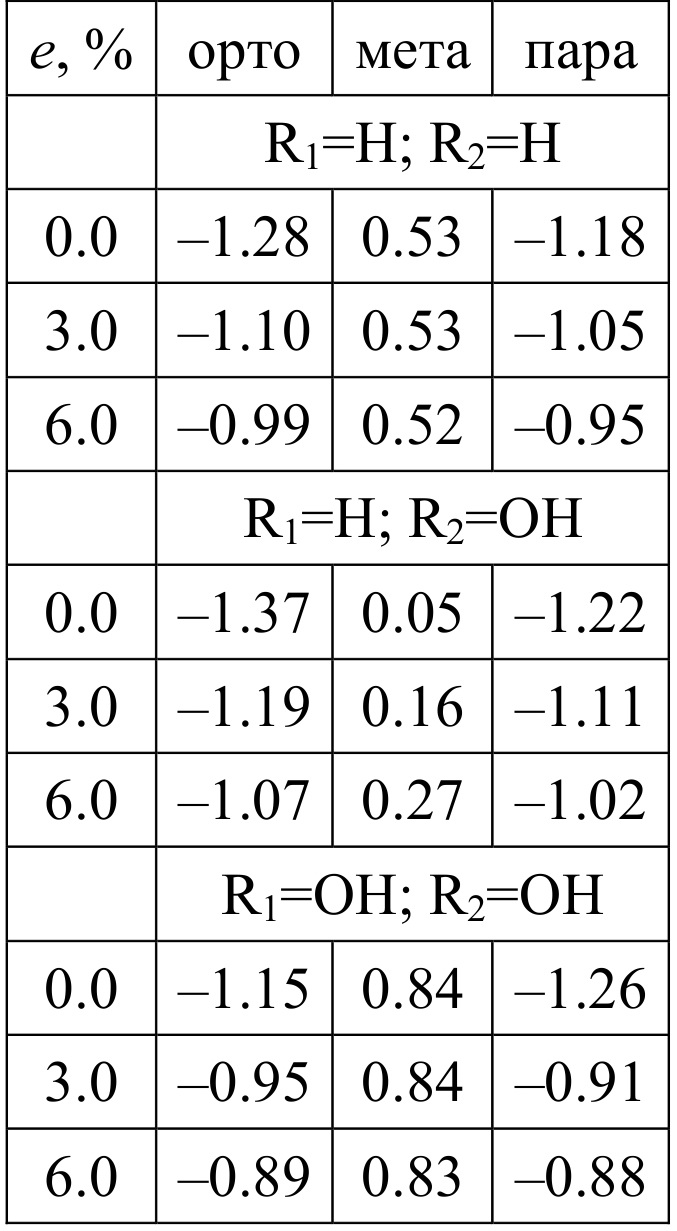
Table 2. Interaction energies Eis (in eV) of two radicals R1 and R2 chemisorbed simultaneously on the same hexagon of free and stretched graphene sheet in ortho, meta and para positions as a function of stretching e (%).
We present a force field molecular dynamics study of nickel-coated graphene sheet bending at temperatures 300 K and 1300 K. Nickel film is represented by nickel atoms located above the centers of carbon hexagons. Parameters for carbon-nickel interaction are fitted with regard to the accurate density functional calculations. Unstrained or flat configuration of the nickel-graphene system is found to be energetically unfavorable for the considered temperatures. Two types of curvatures are taken into account. Positive curvature is characterized by the nickel atoms located closer to the bending axis than the carbon ones, and negative curvature corresponds to the reverse atomic positions. It is found that the equilibrium radius for the curved nickel-graphene complex with the negative curvature is less than the corresponding value for the positive one. However, in both cases the equilibrium curvature radius of nickel-coated graphene is of the order of several nanometers. It is shown that both temperature and bending directions (zigzag and armchair) weakly affect the bending energy and do not change the equilibrium radius. Bending behavior of the system is defined by the carbon-nickel interaction rather than the individual properties of isolated graphene and nickel films.
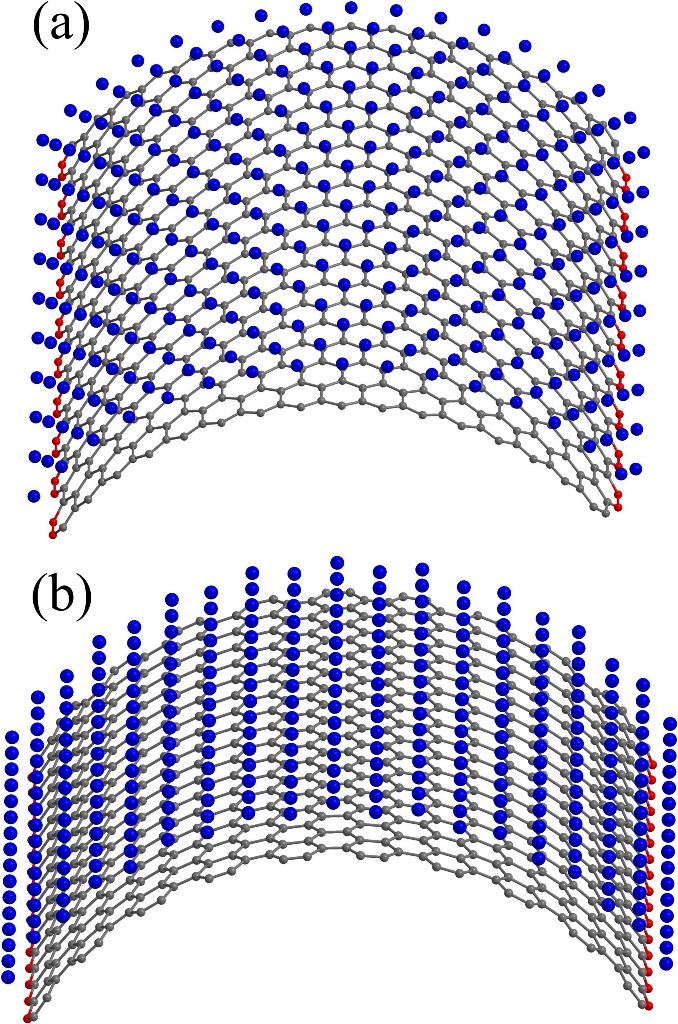
Figure 4. Atomistic model of nickel-coated graphene cluster C678Ni304 with zigzag (a) and armchair (b) curvature. Blue, grey, and red spheres correspond to nickel, free, and fixed carbon atoms, respectively. |
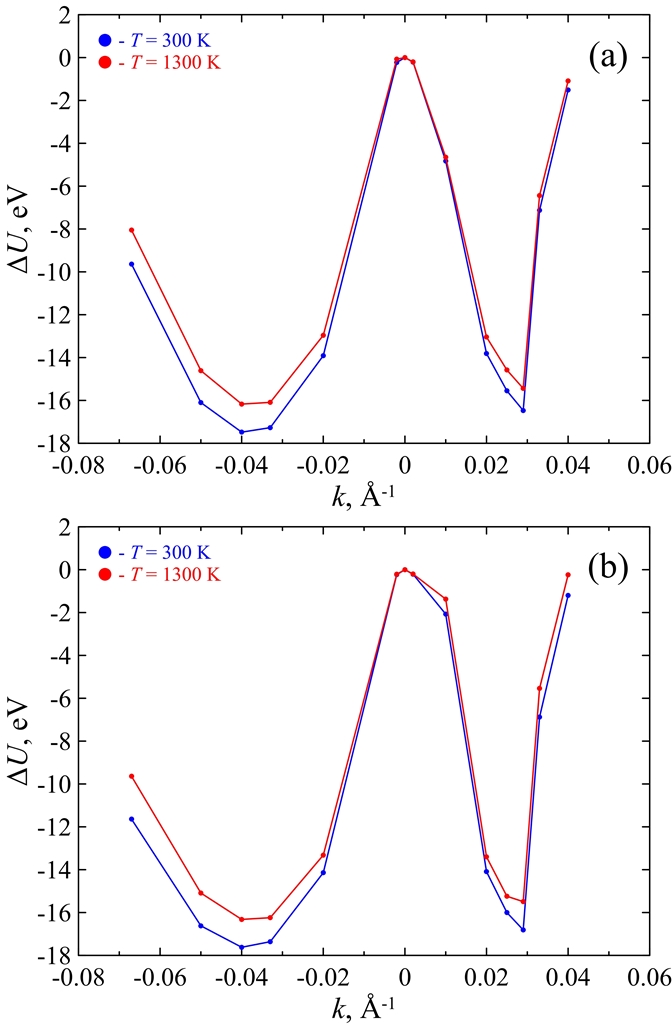
Figure 5. Mean potential energy ΔU of curved C678Ni304 cluster as a function of zigzag (a) and armchair (b) curvature k at T = 300 K (blue) and T = 1300 K (red). The energy of unstrained (i.e., flat) cluster is taken as a reference point. |
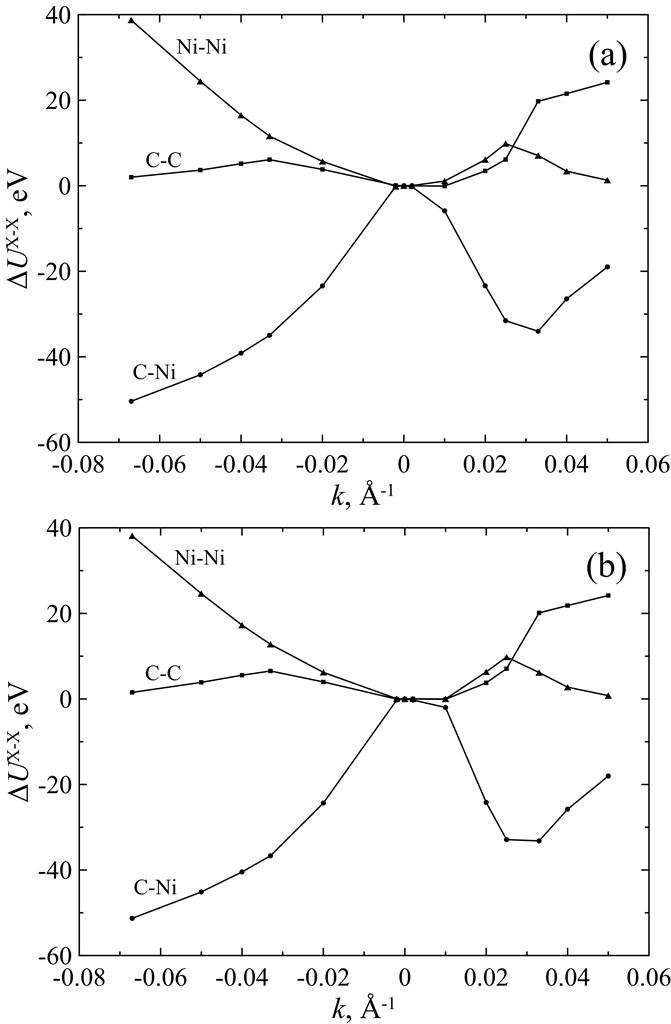
Figure 6. Mean potential energy components ΔUX-X (X = C, Ni) of curved C678Ni304 cluster as a function of zigzag (a) and armchair (b) curvature k at T = 300 K. The energy components of unstrained (i.e., flat) cluster are taken as reference points. |
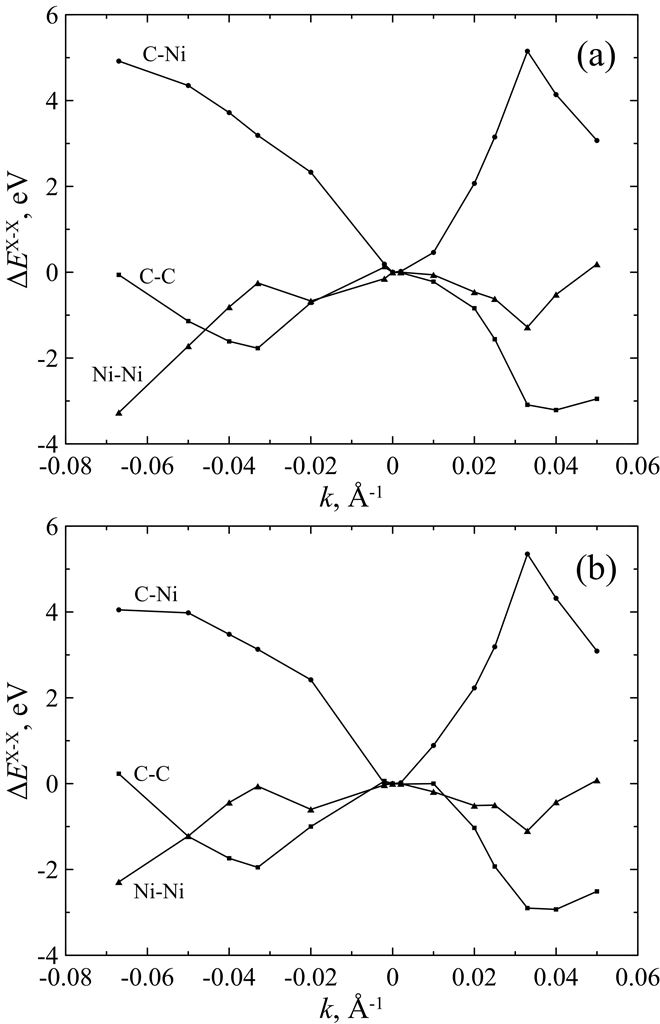
Figure 7. Differences ΔEX-X (X = C, Ni) of mean potential energy components ΔUX-X at T = 1300 K and T = 300 K for curved C678Ni304 cluster as a function of zigzag (a) and armchair (b) curvature k. The energy components ΔUX-X of unstrained (i.e., flat) cluster are taken as reference points. |
We present a density functional study of various polymorphs of graphene fully functionalized with hydrogen, chlorine, fluorine, and hydroxyl groups. We consider the influence of the constant electric field, sheet stretching and compression-induced ripples on relative stabilities of different polymorphs. We found that the effect of the electric field was negligible for all systems. Under mechanical distortions, chlorographene polymorphs decompose, whereas the graphanes, fluorinated and hydroxylated graphenes conserve their identities. Stretching results in moderate decreasing of the energy difference between the common chair-like polymorphs and other structures, but this is insufficient for obtaining the non-common polymorphs. Compression-induced ripples make non-common polymorphs of graphane as well as fluorinated and hydroxylated graphene as thermodynamically stable as the common ones. The ways of experimental observations of non-common polymorphs formed under compression are also discussed.
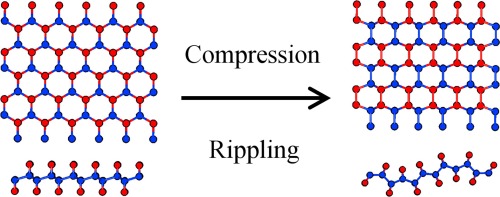
Figure 8. Strained-induced rippling of graphene sheet results in the changing of prevalent ordering type for hydrogen atoms.
Publications
For detailed information see the following publications:
1. Prudkovskiy V.S., Katin K.P., Maslov M.M., Puech P., Yakimova R., Deligeorgis G Efficient cleaning of graphene from residual lithographic polymers by ozone treatment // Carbon. 2016. V. 109. P. 221-226.
2. Katin K.P., Prudkovskiy V.S., Maslov M.M. Chemisorption of hydrogen atoms and hydroxyl groups on stretched graphene: A coupled QM/QM study // Physics Letters A. 2017. V. 381. P. 2686-2690.
3. N.V. Novikov, M.M. Maslov, K.P. Katin, V. S. Prudkovskiy. Effect of DFT-functional on the energy and electronic characteristics of carbon compounds with the unconventional geometry of the framework // Letters on Materials. 2017. Vol. 7(4). P. 433-436.
4. Katin K.P., Prudkovskiy V.S., Maslov M.M. Molecular dynamics simulation of nickel-coated graphene bending // Micro&Nano Letters. 2018. Vol. 13(2). P. 160-164.
5. Dolinskii, I.Y., Katin, K.P., Grishakov, K.S., Prudkovskiy V.S., Kargin, N.I., Maslov, M.M. Influence of Mechanical Stretching on Adsorption Properties of Nitrogen-Doped Graphene // Physics of the Solid State. 2018. V. 60(4). P. 821-825.
6. Prudkovskiy V.S., Katin K.P., Maslov M.M., Puech P., Yakimova R., Deligeorgis G. High robustness of epitaxial 4H-SiC graphene to oxidation processes // J. Phys.: Conf. Ser. 2018. V. 1124. P. 081020.
7. Prudkovskiy, V.S., Iacovella, F., Katin, K.P., Maslov, M.M., Cherkashin, N. A bottom-up approach for controlled deformation of carbon nanotubes through blistering of supporting substrate surface // Nanotechnology. 2018. V. 29(36). P. 365304.
8. Grishakov K.S., Katin K.P., Prudkovskiy V.S., Maslov M.M. Relative stabilities of various fully functionalized graphene polymorphs under mechanical strain and electric field // Applied Surface Science. 2019. V. 463. P. 1051-1057.
9. I.Yu. Dolinskii, K.S. Grishakov, V.S. Prudkovskii. Effect of a Nitrogen Doping and a Mechanical Stress on the Adsorption Capacity of Graphdiene // Physics of the Solid State. 2019. Vol. 61(2). P. 274–278.
Acknowledgements
The reported study was funded by RFBR, according to the research project No. 16-32-60081 mol_a_dk.