
Energy-efficient nitrogen-containing atomic systems and nanomaterials based on them for the new generation energy sources
This webpage contains the main results of computer simulation of pure nitrogen and nitrogen-containing high-energy-density materials obtained in the framework of the scientific project “Energy-efficient nitrogen-containing atomic systems and nanomaterials based on them for the new generation energy sources” that is supported by the Russian Foundation for Basic Research (RFBR) (Grant No. 18-32-20139 mol_a_ved).
Why is nitrogen the fuel of the future?
A significant amount of energy can be released in the decomposition of metastable nitrogen-containing atomic clusters due to the formation of the N2 molecules. So for the pure nitrogen clusters energy-release exceeds 2 eV/atom (energy of the system is reduced greatly due to the rupture of single covalent N–N bonds and the formation of triple N–N bonds). For this reason, nitrogen-containing atomic clusters are regarded as the functional units of high-energy materials and next-generation fuels of high-density energy stored (HEDM – high-energy-density materials).
What is needed to be done to accelerate the dissemination of nitrogen as fuel?
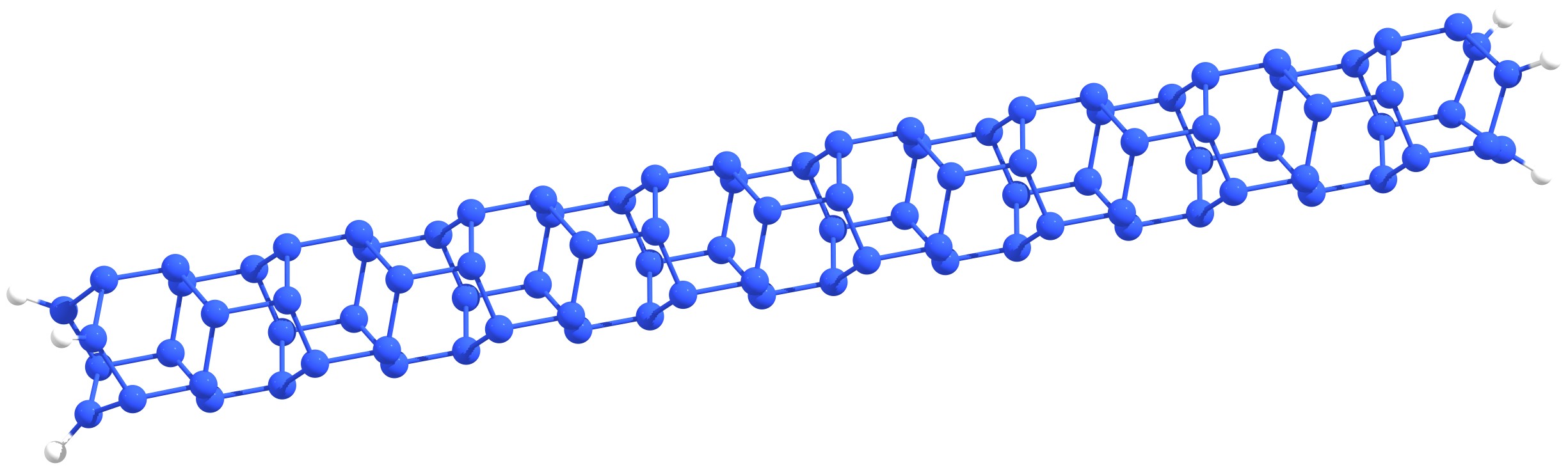
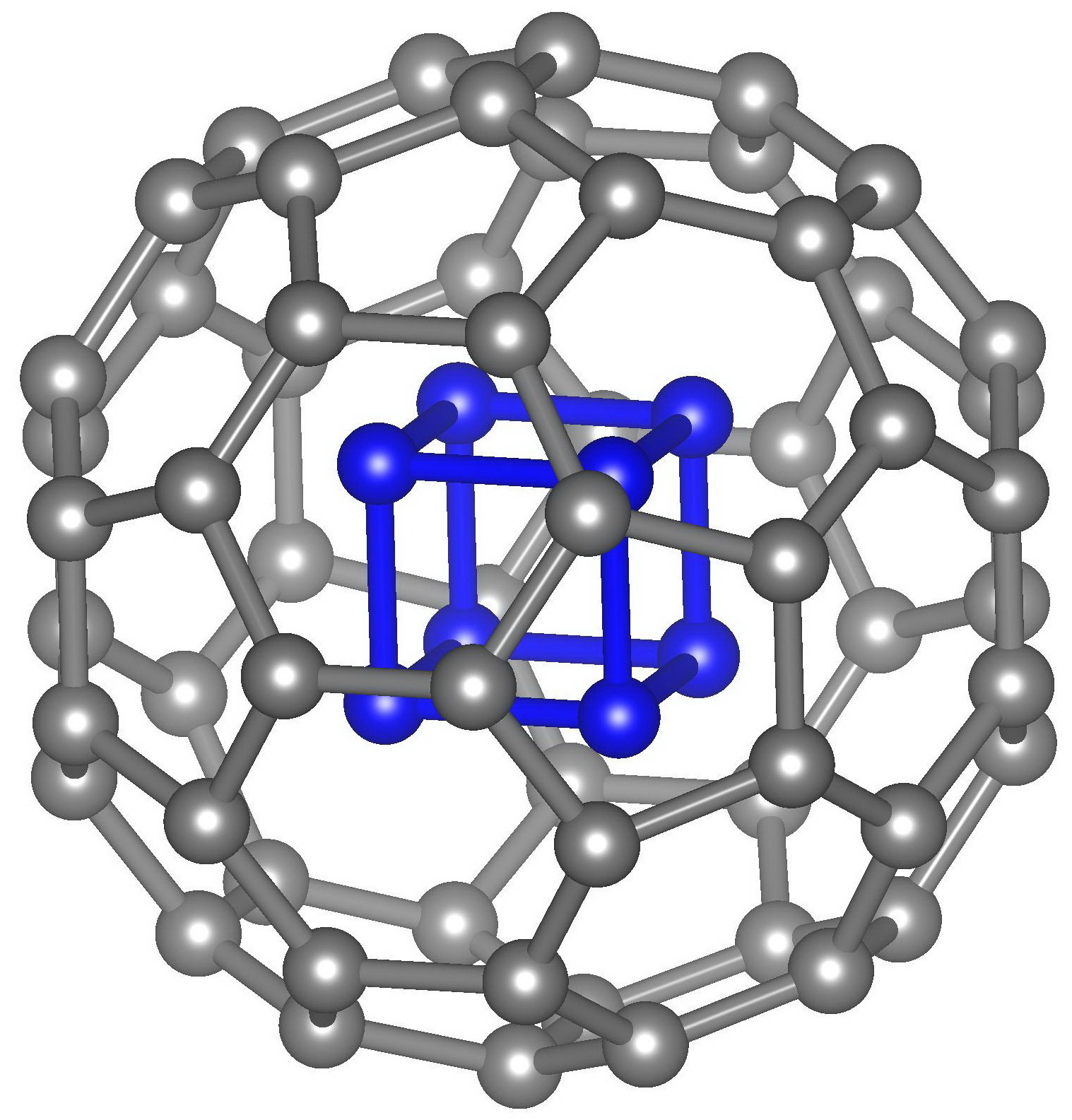
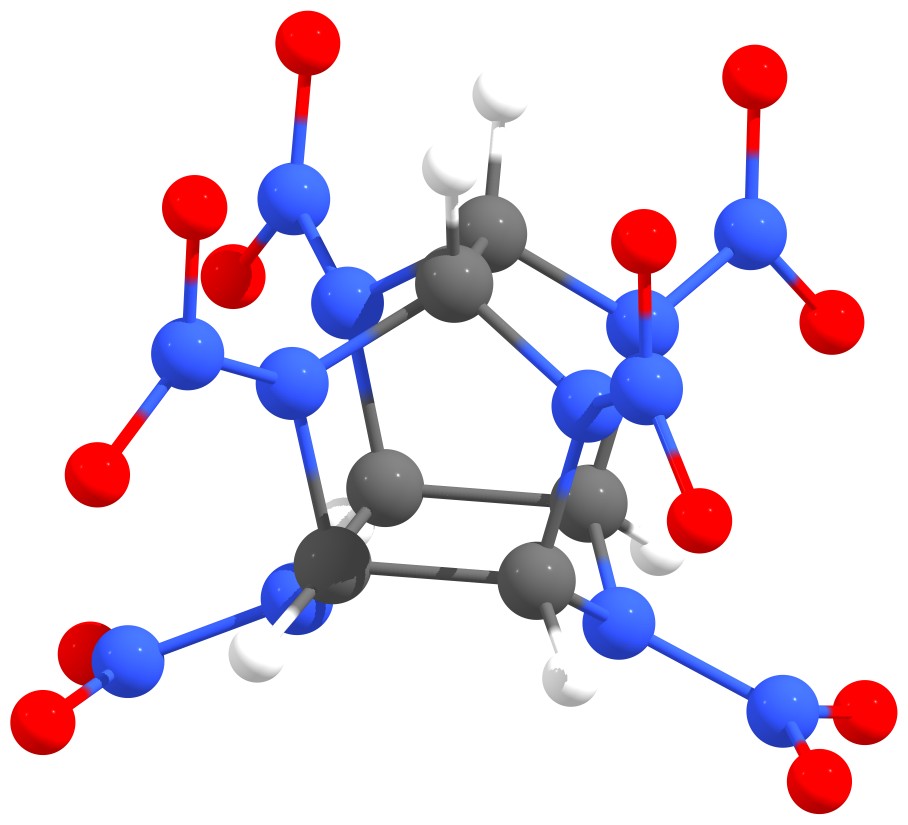
Despite the intensive research stimulated by the successful synthesis of polymeric nitrogen and the number of small charged nitrogen clusters and nanomaterials based on them in early 2000s, until now the pure nitrogen structures, which are stable at atmospheric pressure and room temperature are not obtained. At the same time, some high-energy organic clusters containing in addition to nitrogen carbon, oxygen and hydrogen atoms (e.g., CL-20, polynitrocubane, etc.) exhibit high thermal stability. Therefore, proper nitrogen stabilization is the key to its dissemination as a fuel. Stabilization can be carried out in different ways: a) the boundary/surface doping (passivation), b) the formation of endohedral complexes (the introduction of pure nitrogen clusters inside the carbon cage), and c) the creation of the combined carbon/silicon-nitrogen framework. Examples are shown in Figure 1.
|
|
(a) |
|
|
(b) |
|
|
(c) |
|
Figure 1 – Examples of nitrogen stabilization: nitrogen nanotube edge doping with hydrogen atoms (a), N8@C60 endohedral complex (b), carbon-nitrogen CL-20 framework (c). |
Structural features indicating the stability of nitrogen cages
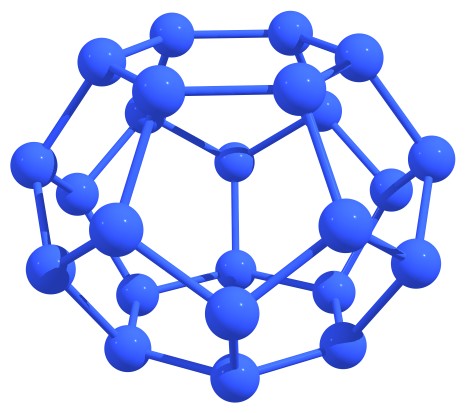
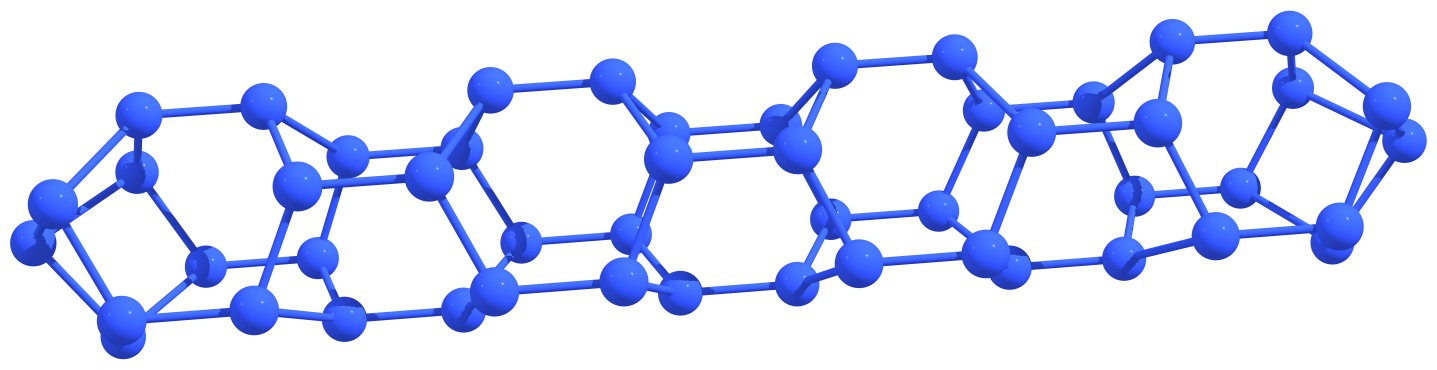
After the traditional carbon fullerenes were discovered, the isolated-pentagon rule (IPR) was proposed. This rule can be formulated as follows: the most stable carbon fullerenes are those in which no two pentagons share an edge, that is, each pentagon is completely surrounded by hexagons. Unfortunately, for nitrogen fullerenes, this rule turned out to be inapplicable. We were able to note a number of topological features that may indicate the stability of nitrogen cages or quasi-fullerenes. So, stable structures are those in which six-membered nitrogen rings (hexagons) are absent or maximally distant from each other and/or nitrogen atoms in adjacent hexagons do not lie in the same plane, forming (due to concavity) the so-called “nitrogen boats”. The first case includes N20 or N24 fullerenes, and the last case includes the extended nitrogen nanotubes, see Figure 2.
|
|
|
(a) |
|
|
|
|
(b) |
|
|
Figure 2 – Stable nitrogen fullerenes N20 (left) and N24 (right) (a) and capped nanotube (b) with characteristic structural features indicating the stability. |
|
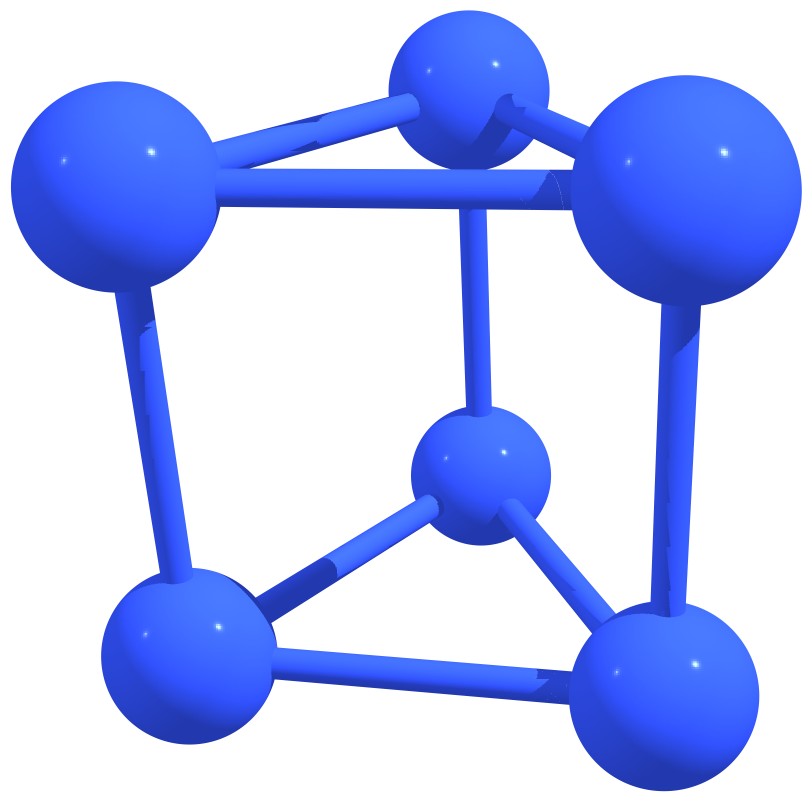
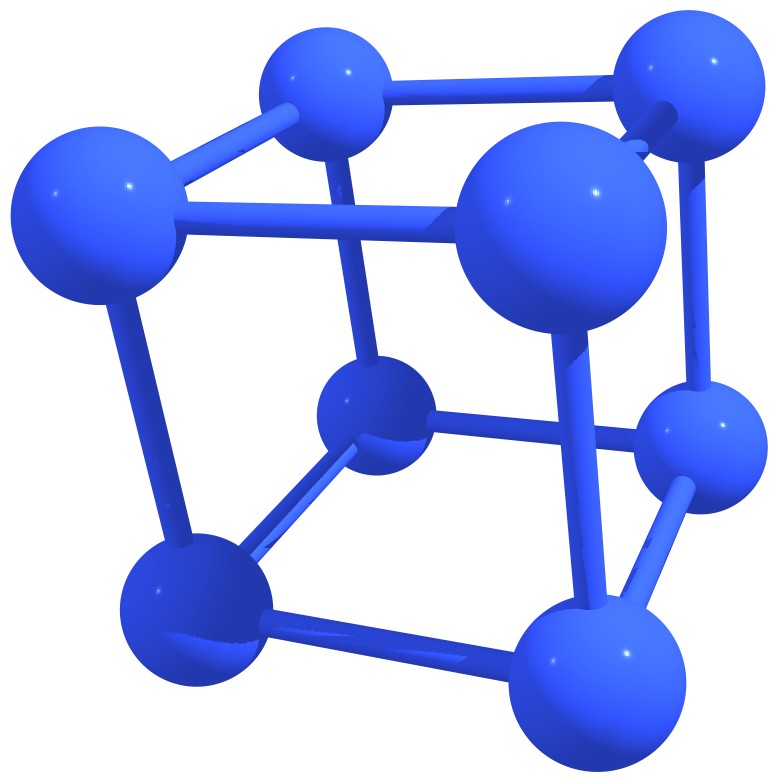
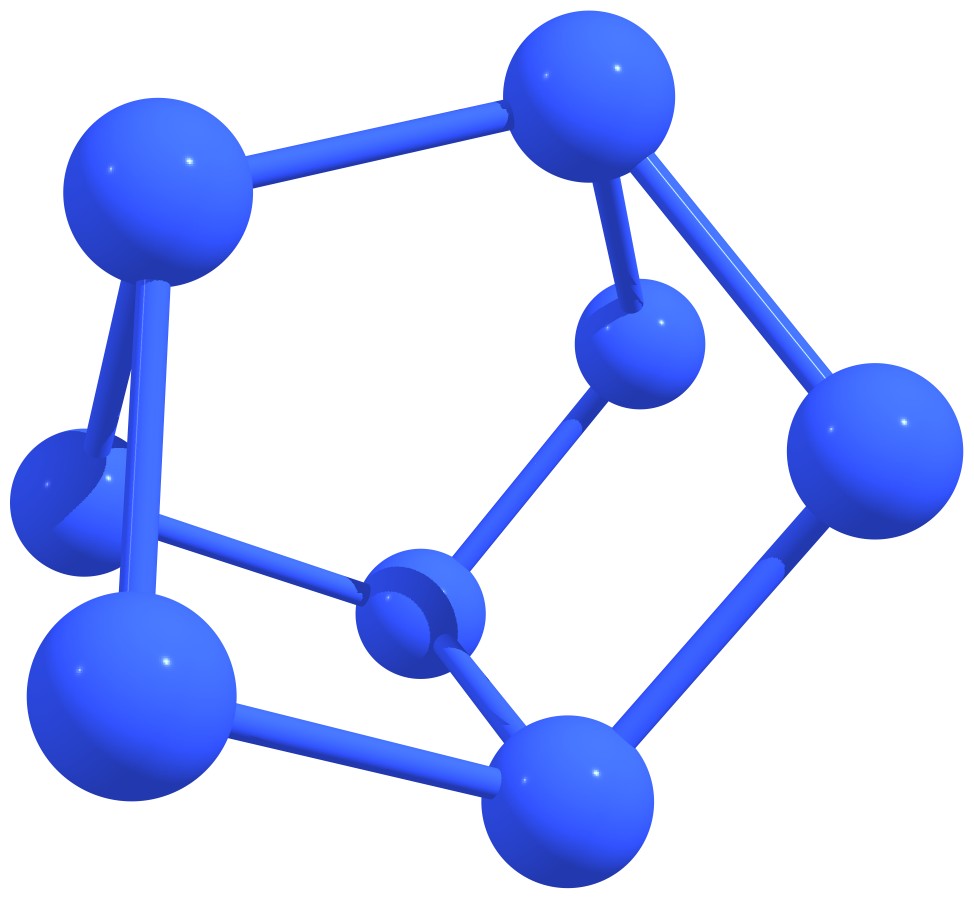
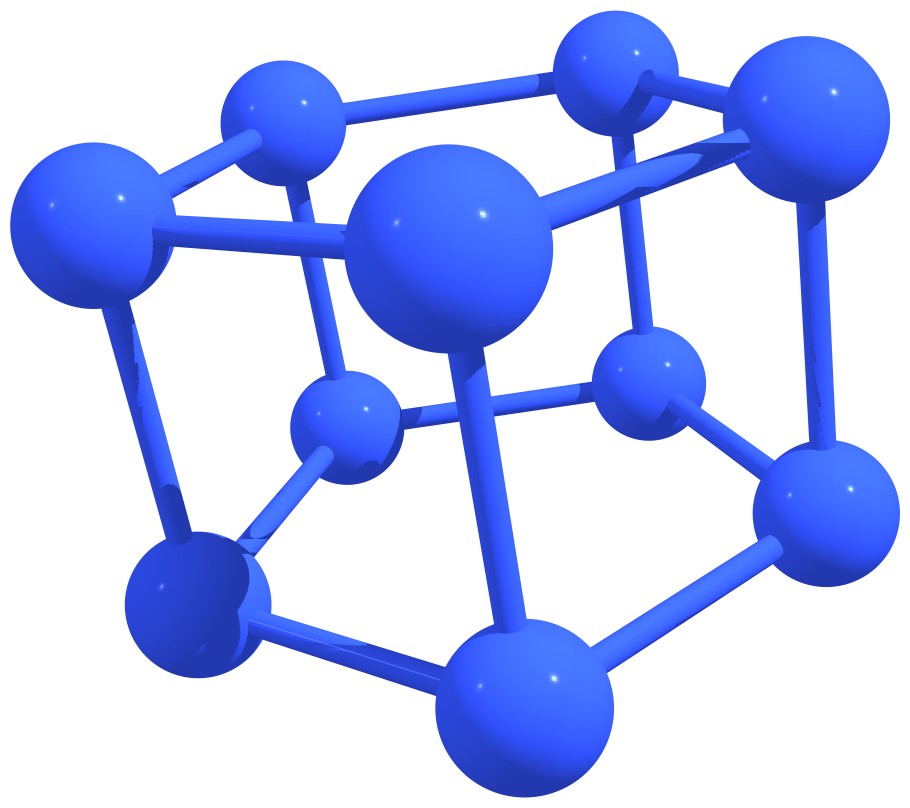
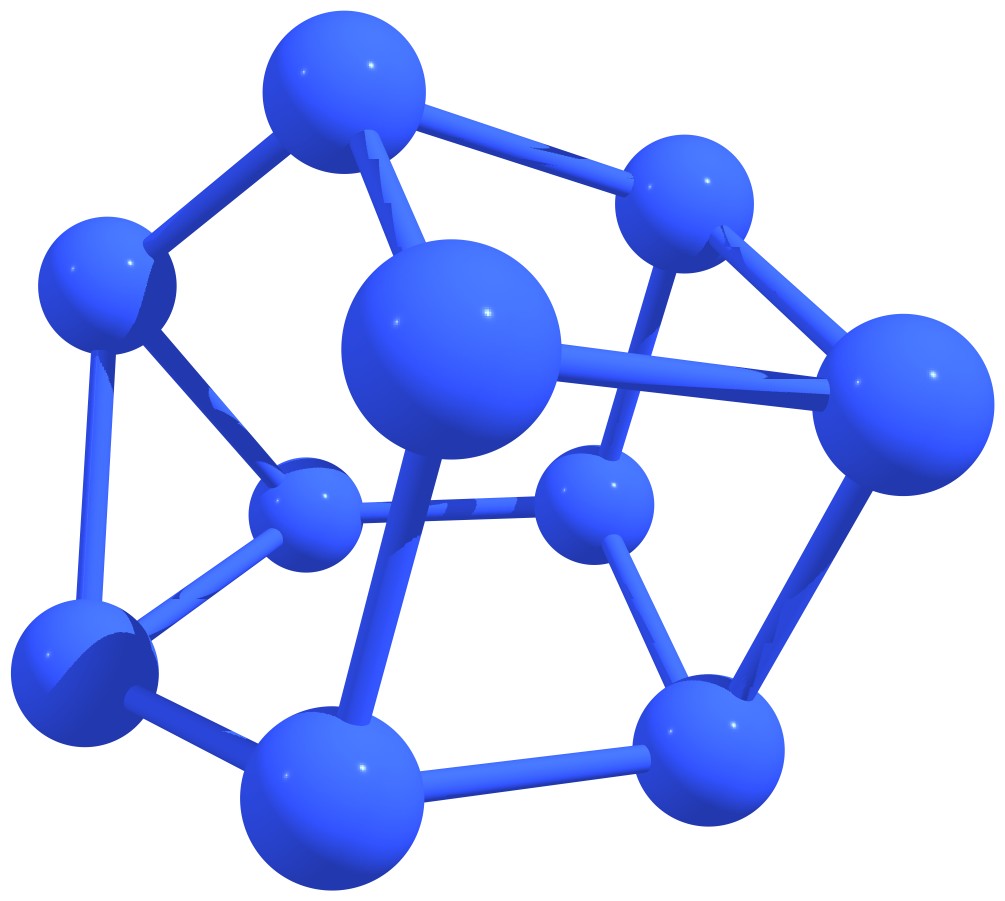
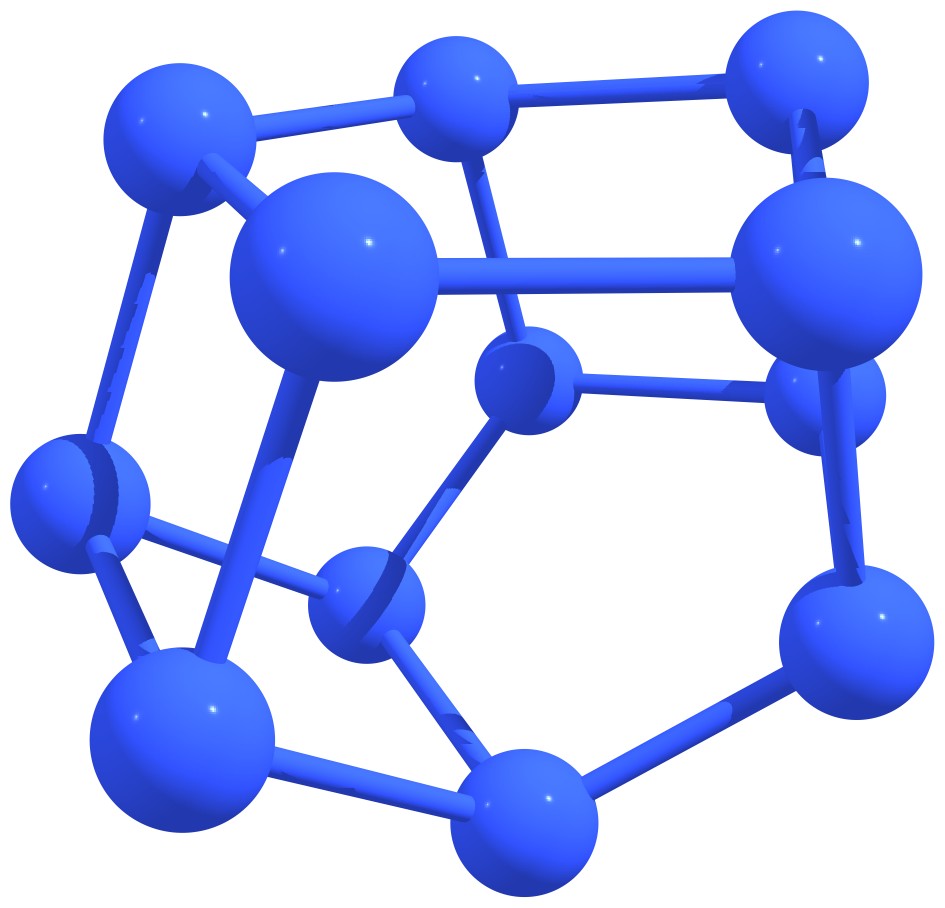
Stable nitrogen quasifullerens are presented in Figure 3.
|
|
|
|
(a) |
(b) |
(c) |
|
|
|
|
(d) |
(e) |
(f) |
|
|
|
|
(g) |
(h) |
(i) |
|
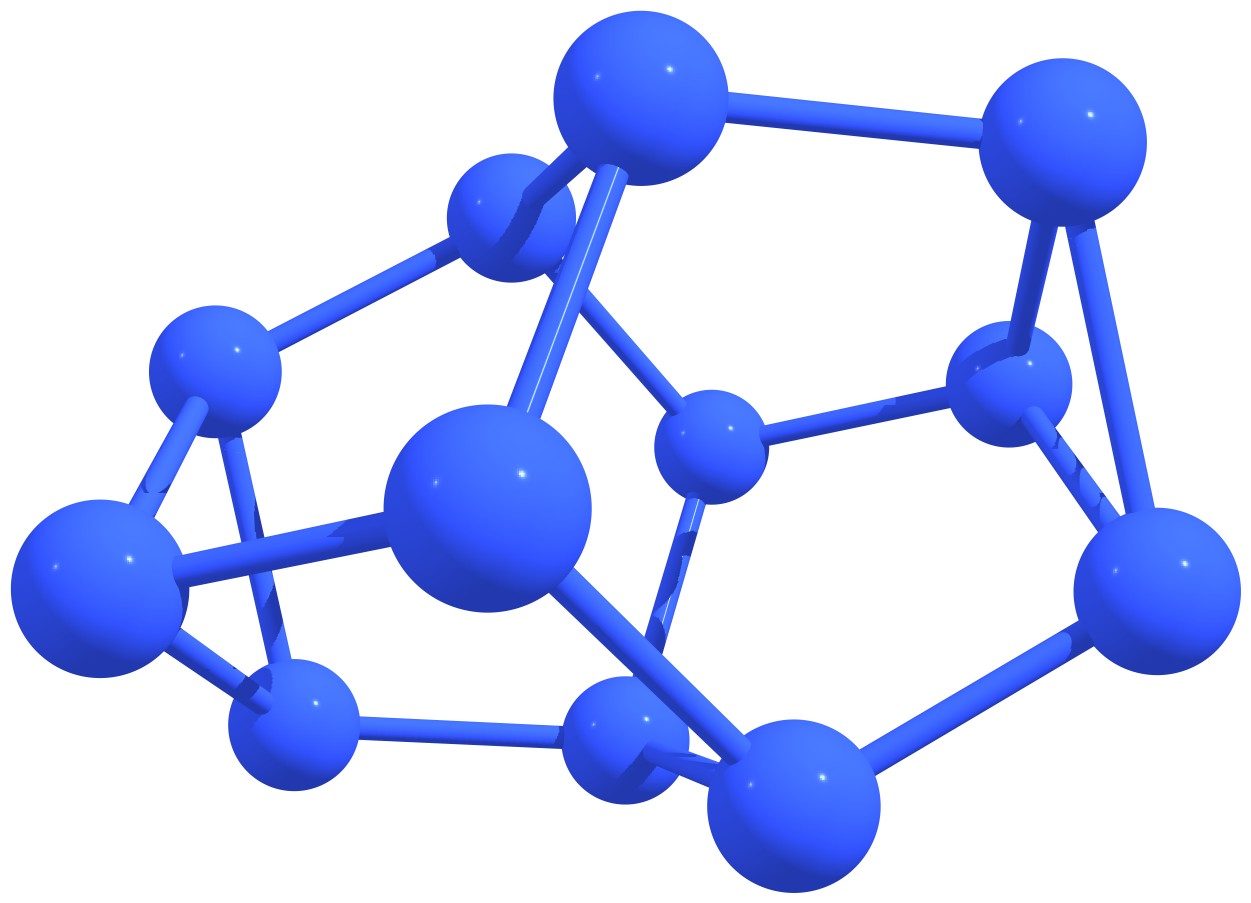
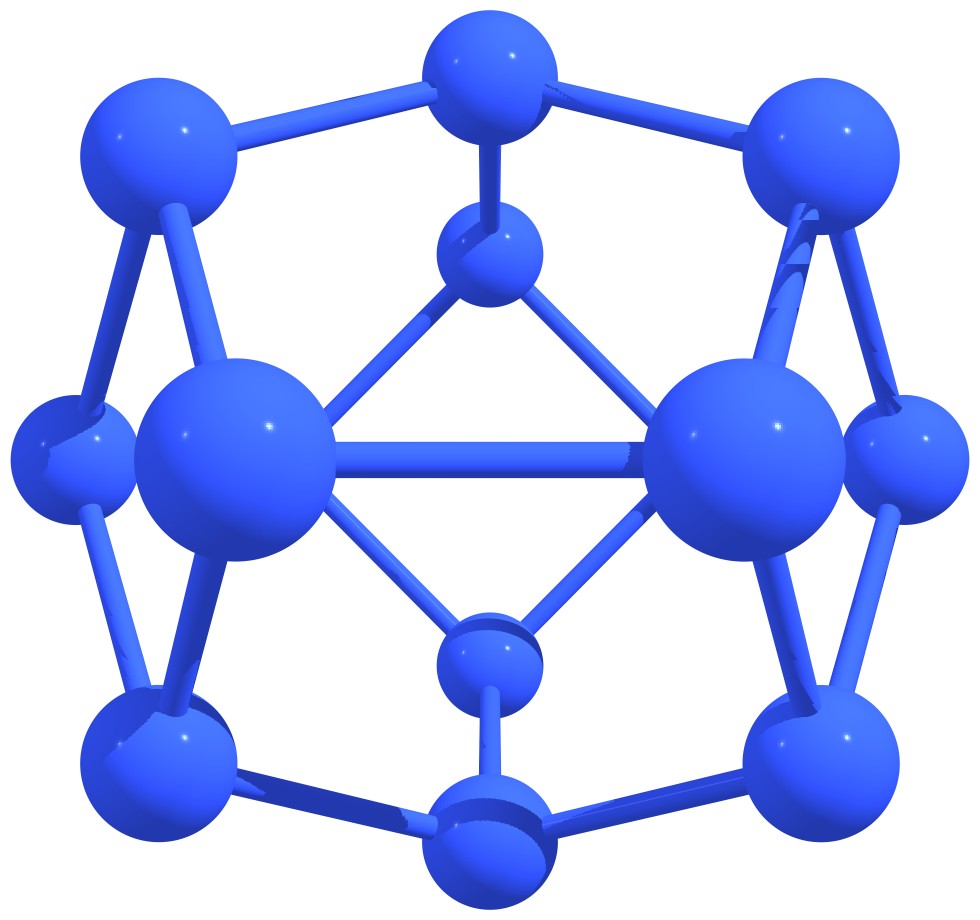
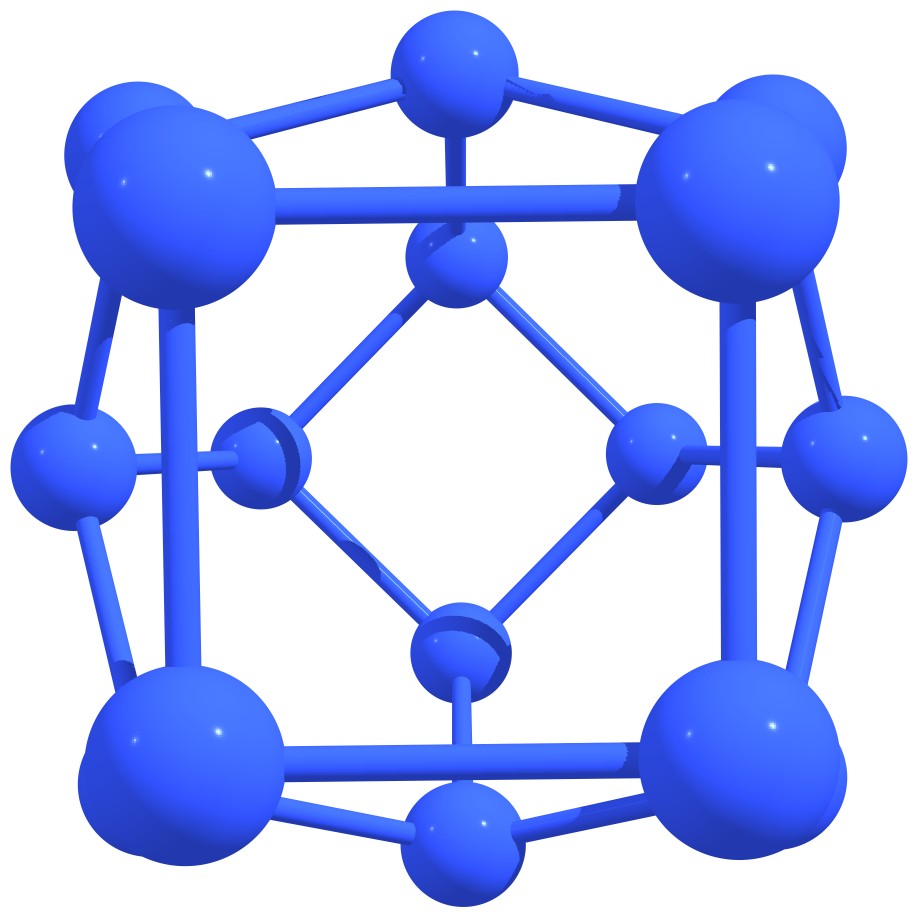
Figure 3 – Stable nitrogen quasi-fullerenes: N6 (a), N8 (b), N8 (c), N10 (d), N10 (e), N12 (f), N12 (g), N14 (h), and N16 (i). |
||
Nitrogen nanotubes
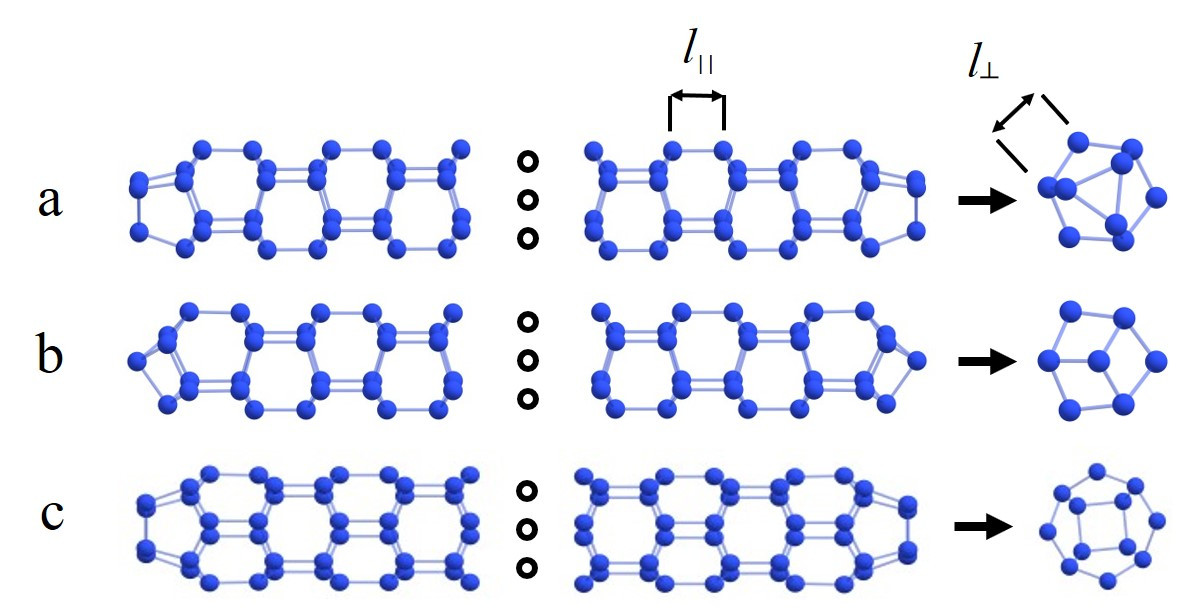
We apply the density functional theory to investigate structural, energetic, and electronic properties and stability of extended armchair and zigzag nitrogen nanotubes with a length of ≈ 3 nm. The capping effect, as well as the passivation of nanotubes’ ends by hydrogen atoms and hydroxyl groups on their stability, are studied. According to our calculations, pristine nitrogen nanotubes are unstable. Both capping and passivation of the nanotube end provide thermodynamic stability only for (3,0) and (4,0) zigzag nitrogen nanotubes (see Figure 4). Moreover, the calculated frequency spectra of considered systems confirm their dynamic stability. The calculated HOMO-LUMO gaps for these stable extended systems are of the order of 4 eV, so they can be assigned to the class of insulators. It is shown that nitrogen nanotubes are able to store a large amount of energy and can be used as a basis for new high-energy-density materials.
|
|
Figure 4 – Atomic structures of the capped nitrogen nanotubes (a) (3,0)NNT(N102), (b) (3,0)NNT(N98), (c) (4,0)NNT(N136). Top-side view and bottom-top view. Symbols l|| and l┴ correspond to the interlayer and intralayer N-N bonds in the distance of the capped ends, respectively. |
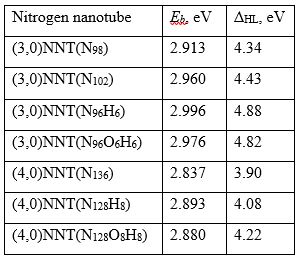
Table 1 presents the values of HOMO-LUMO gaps ΔHL and binding energies Eb.
|
|
Table 1. Binding energies (Eb) and HOMO-LUMO gaps (ΔHL) for the stable nitrogen nanotubes. |
To estimate the stored energy in our stable nitrogen nanotubes, we determine the reaction energy for one atom
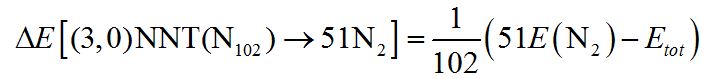
is equal to 1.88 eV/atom. This value is comparable with the calculated energy difference ΔE[N4→2N2]=2 eV/atom for experimentally synthesized N4, which demonstrates the prospects of using these systems as the high-energy-density materials (Figure 5).
|
|
Figure 5 – Schematic illustration of energy release during the nitrogen nanotube decomposition into the molecular nitrogen. |
So, only (3,0) and (4,0) zigzag nitrogen nanotubes with extremely small diameters can be stable under ambient conditions. Their stability can be achieved via the capping or passivation of their ends by the hydrogen atoms or hydroxyl groups. Among all the systems considered, the most energetically feasible is the (3,0) nitrogen nanotube with passivated by hydrogen atoms ends. It can release a large amount of energy during the decomposition to the isolated nitrogen molecules.
Nitrogen astralenes
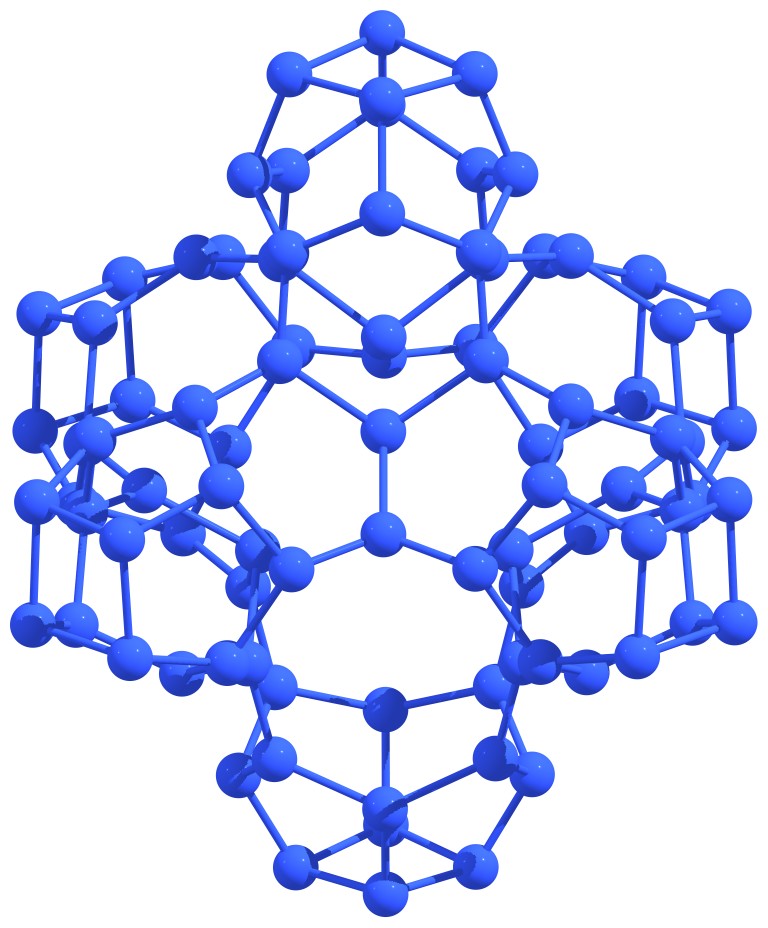
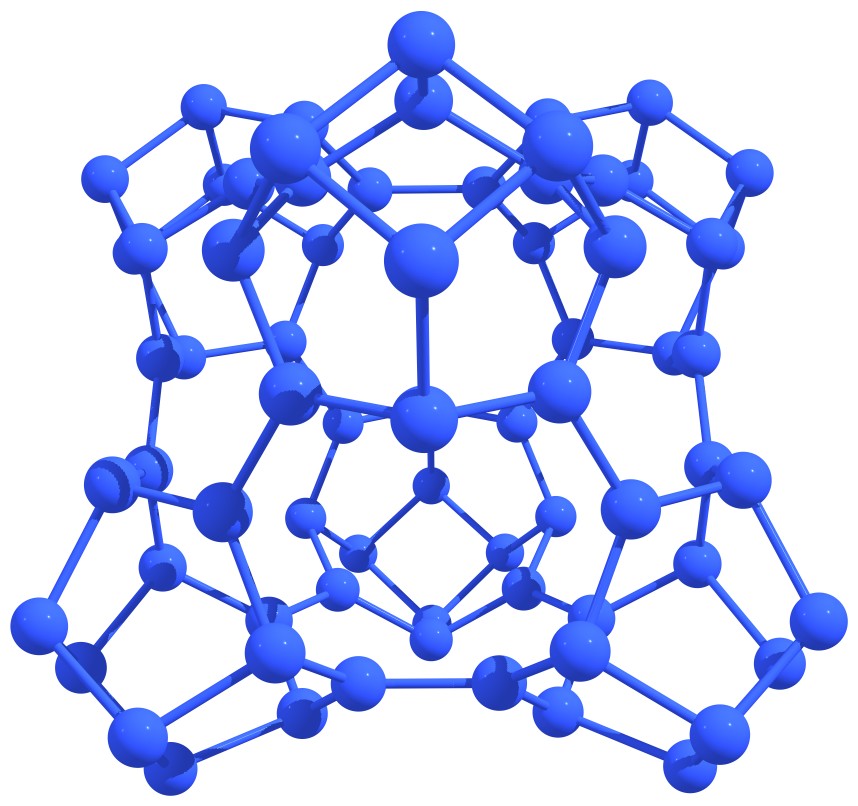
Nitrogen astralenes is a new class of purely nitrogen systems. Nitrogen astralenes are the complex molecular covalent forms built from the fragments of nitrogen nanotubes and nitrogen fullerenes. Examples of nitrogen astralenes are presented in Figure 6.
|
|
|
Figure 6 – Examples of elementary nitrogen astralenes. |
|
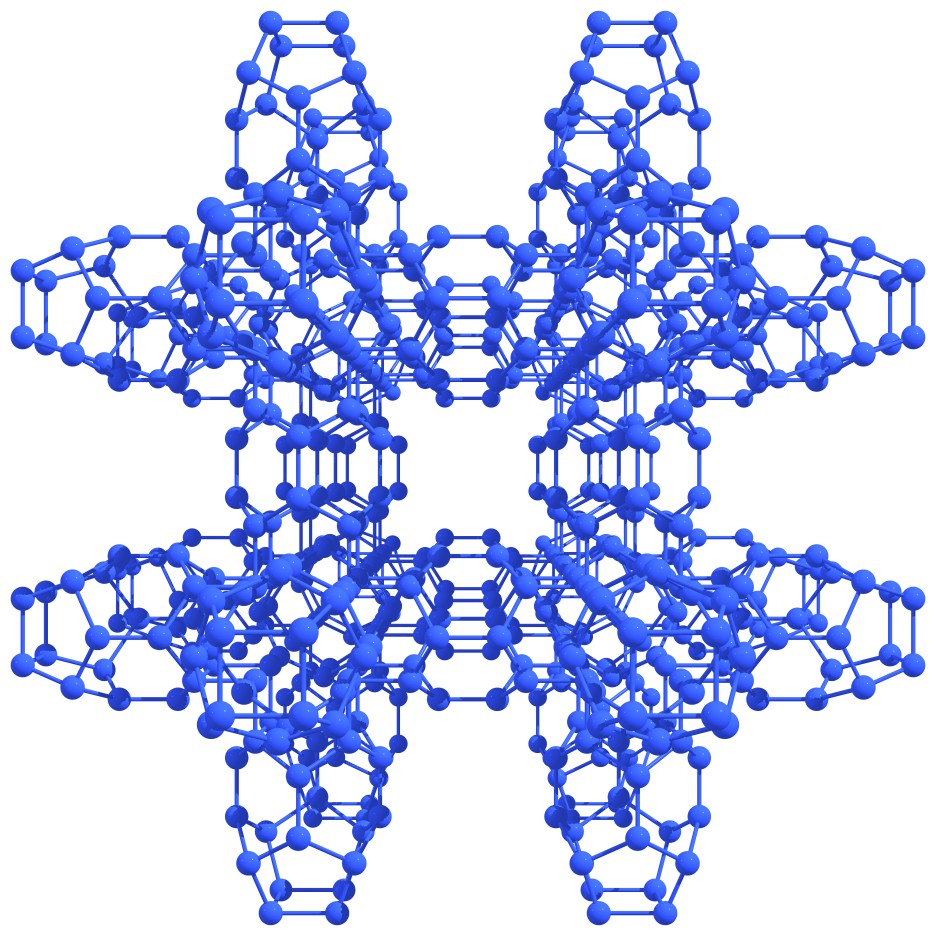
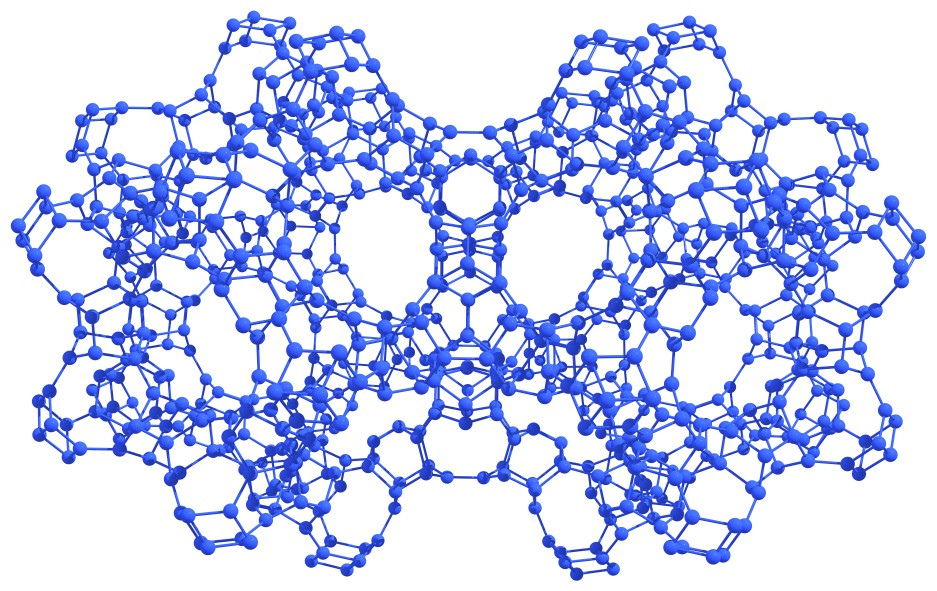
On the basis of nitrogen astralenes, the formation of a wide variety of covalent high-energy crystals is possible through the direct covalent bonding of isolated astralenes (see Figure 7).
|
|
|
Figure 7 – Fragments of nitrogen covalent crystals based on the nitrogen astralenes. |
|
Cubane inside the fullerene
Within the framework of this project, we analyze how the C60 shell affects the stability of the nitrogen cubane system N8 placed inside. Cubic N8 system is the all-nitrogen analogous of well-known hydrocarbon cubane C8H8, which is the basis of the broad family of the already synthesized high-energy-density materials. The N8 effective size is a reasonable compromise between rather small highly strained systems (N4, N6) and too large clusters (N10, N12), which is quite difficult to place inside the C60. The cubic structure of N8 was found to be stable: its lowest vibrational frequency by some estimates was about 530 cm-1. Because of its strained structure, N8 cage possesses higher total energy than the other nine possible N8 isomers. On the other hand, the relative energies of different competitive isomers can change under the action of spatial constraints. Cubic system N8 has the smallest effective diameter among all N8 isomers, and its shape and size are very suitable for the encapsulation inside the nearly spherical C60.
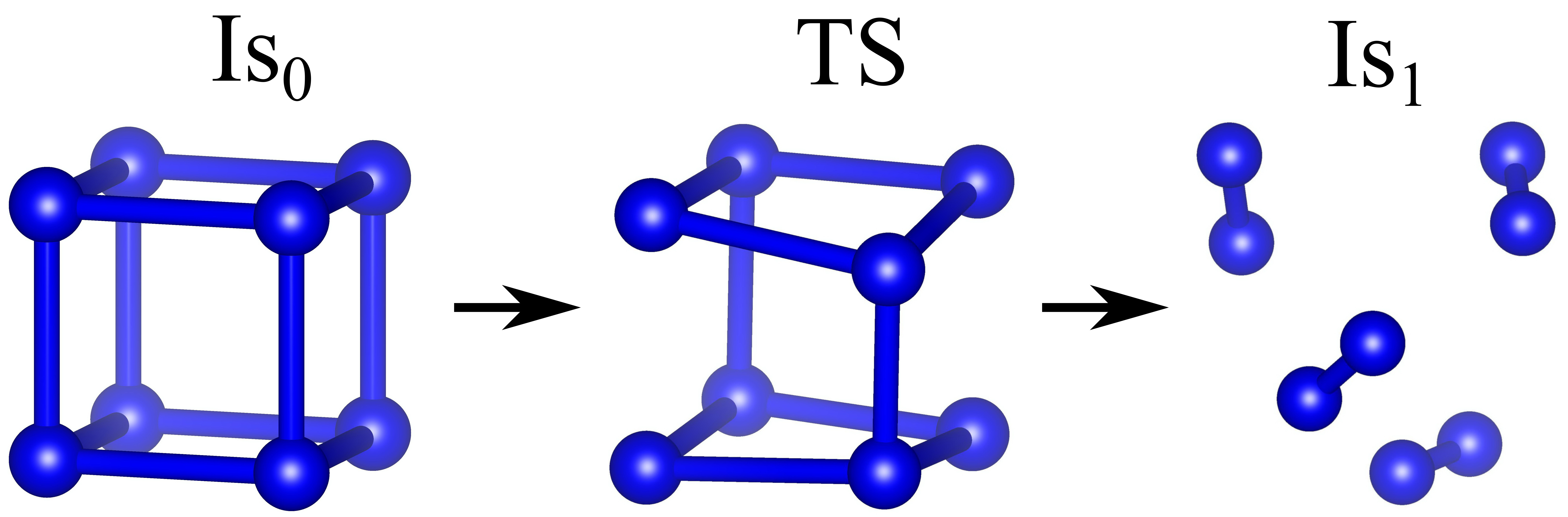
Using the original method of finding the saddle configuration implemented in the NTBM program (ntbm.info), we are able to determine the decomposition scheme of the isolated cubane N8 and cubane N8 inside the C60 cage, as well as to calculate the minimum energy barrier U preventing the caged cubane decomposition. Figure 8 presents the isolated N8 decomposition path, and Figure 9 shows the N8 decomposition path inside the fullerene cage.
|
|
Figure 8 – The mechanism of the isolated cubane N8 decomposition. Is0 is the initial configuration corresponded to the energy minimum, TS is the transition state or saddle point, and Is1 is the decomposition product represented by the nitrogen molecules. |
|
|
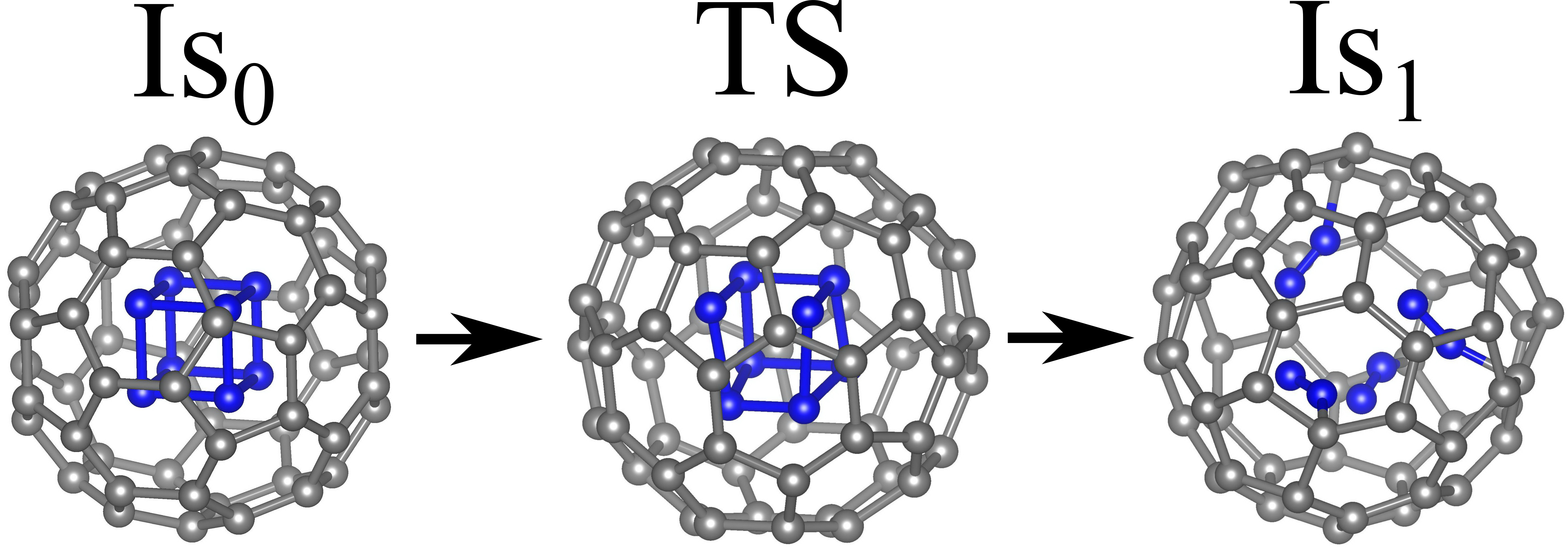
Figure 9 – The mechanism of cubane N8 decomposition inside the fullerene C60. Is0 corresponds to the undistorted configuration cubane@C60, TS is the transition state or saddle point, and Is1 is the decomposition product represented by the nitrogen molecules attached to the inner surface of the fullerene cage. |
The kinetic stability of the system is determined by the height of the minimum energy barrier separating Is0 from Is1. The detailed analysis shows that the lowest energy path of both isolated and caged cubane N8 rearrangement leads to the molecular nitrogen through the single saddle configuration (see Figures 8 and 9). However, in the case of caged N8, the nitrogen molecules N2 can be covalently bound with the inner surface of the fullerene cage (Figure 9). According to our calculations, the value of U equals 0.11 eV for the isolated cubane molecule and 0.20 eV for the caged one. It should be noted that the energy barrier U for the encapsulated cubane is twice as large as the corresponding value for the isolated one. Thus, the fullerene cage significantly increases the stability of the N8 cube.
To analyze the kinetic stability of N8 inside the fullerene cage in detail and follow the decay process in real-time, we perform the molecular dynamics simulation for the N8@C60 complex at different temperatures. The activation energy Ea of cubane decomposition is determined by analyzing the temperature dependence of its lifetime τ using the Arrhenius equation. Approximating the inverse temperature dependence of ln(τ) by a straight line, we can determine activation energy Ea and frequency factor A by the slope angle of this line and its y-intercept, respectively. These values and their standard deviations are found to be Ea=0.18±0.01 eV and A=1015.23±0.37 s-1.
The molecular dynamics data obtained allowed us to find the temperature dependence of the caged N8 lifetime and to determine the activation energy and the frequency factor for its decay. These data are confirmed by the transition state search and further calculation of the minimum energy barrier preventing the decay. It is confirmed that the C60 significantly increases the stability of the N8 cube.
CL-20 frameworks
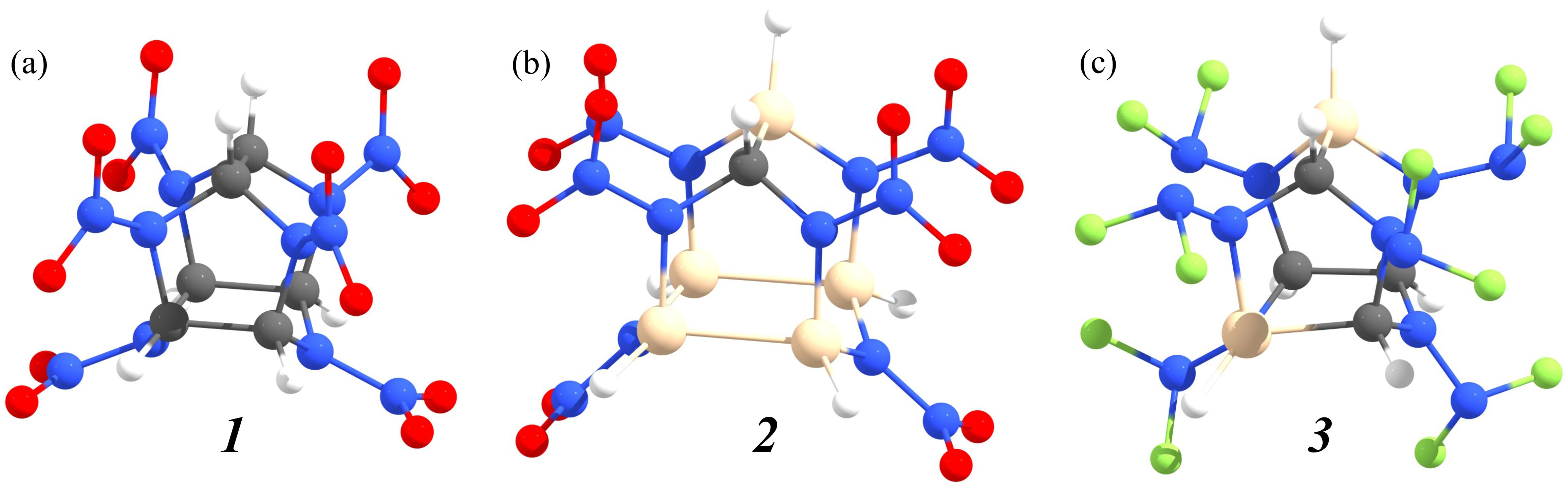
A CL-20 based cages in which carbon/oxygen atoms are replaced by silicon/fluorine ones (Figure 10) are studied using the ab initio molecular dynamics, density functional theory, and time-dependent density functional theory. In contrast to the pristine CL-20, the first step of the pyrolysis of these cages is the migration of oxygen/fluorine atoms to silicon. Molecules containing fluorine are unstable at room temperature. The high-energy silicon-containing molecule (CSi5H6N12O12) is approximately as stable as pristine CL-20. The energy barrier preventing its decomposition is about 200 kJ/mol. Energies of the frontier orbitals and reactivity descriptors of CSi5H6N12O12 are very close to the corresponding values of pure CL-20. All studied cages can form covalent dimers via the methylene molecular bridges. It is found that the reactions of dimerization are exothermic. Dimers’ isomers in which silicon atoms are located closer to the methylene bridges possess lower internal energies. It is found that the mechanisms of dimers’ thermal decomposition are similar to the analog mechanisms of corresponding monomers. The dimerization of the cages results in the redshifts of their ultraviolet spectra.
|
|
Figure 10 – Atomic structures of C6H6N12O12 (CL-20, 1) (a) and its substituted derivatives CSi5H6N12O12 2 (b) and C4Si2H6N12F12 3 (c). Grey, blue, red, white, brown, and green circles correspond to carbon, nitrogen, oxygen, hydrogen, silicon, and fluorine atoms, respectively. |
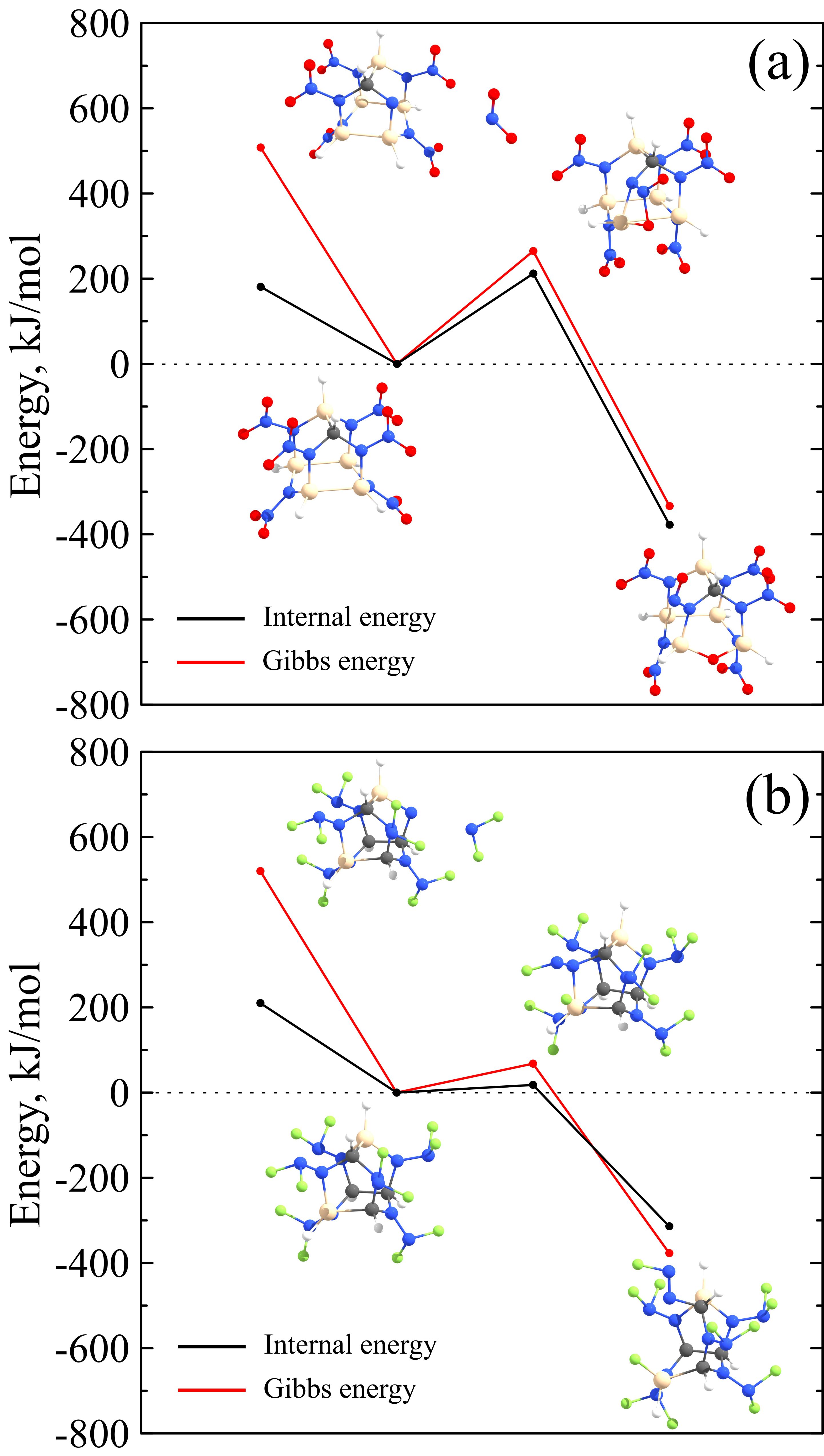
CL-20 is a highly strained metastable compound and, therefore, it decomposes at high temperatures with heat releasing. According to the previous studies, decomposition of CL-20 starts with the fission of the nitro group that induces a skeleton destabilization and further decomposition. So, the NO2 fission determines the overall activation barrier. The N–NO2 bond dissociation is a barrierless process, and the corresponding bond dissociation energy needed for the NO2 fission is about 200 kJ/mol. It was proposed a natural assumption that the same mechanism (NO2 or NF2 fission) was also relevant to the CL-20 derivatives. However, this assumption was not confirmed by our ab initio molecular dynamics simulations. Observing the evolution of heated CL-20 derivatives, we found two competitive mechanisms of the initial decomposition step. The first mechanism is the NO2 or NF2 fission, and the second one is a migration of O or F atom with the formation of Si–O or Si–F covalent bond. Migration is possible for the Si-containing CL-20 derivatives because silicon possesses a larger covalent radius (1.11 Å) than the carbon (0.76 Å). Corresponding energetic diagrams are presented in Figure 11.
|
|
Figure 11 – Two competitive mechanisms of 2 (a) and 3 (b) decomposition: NO2/NF2 fission versus O/F migration to Si. Black and red lines correspond to the internal energies and Gibbs energies at T = 3000 K, respectively. |
For the 2 system, the barrier for the NO2 fission is somewhat lower than that for the O migration. Therefore, the NO2 fission mechanism prevails at room temperature. However, at higher temperatures, one should consider thermal corrections. For example, at T = 3000 K (it is about the half of CL-20 explosive temperature) O migration mechanism becomes more feasible with regard to the lower Gibbs energy, as it is presented in Figure 11a. Anyway, the energy barrier for the CSi5H6N12O12 decomposition is about 200 kJ/mol. This value is close to the corresponding value for the pristine CL-20. Therefore, it can be said that 2 is as kinetically stable as pristine CL-20. In contrast to oxygen, fluorine is able to easily migrate to the silicon atom and to destabilize the 3 cage, see Figure 11b. So, the 3 system is much less stable. To evaluate its lifetime t before the decomposition, we adopt the Arrhenius formula. In our evaluation, we assume the activation energy is equal to the energy barrier (0.18 eV, see Figure 11b). Frequency factor w can be defined from the Vineyard formula. So, we obtain Ea = 0.18 eV and w = 1.93·1015 1/s. In accordance with the Arrhenius formula, t ~ 0.5 ps at T = 300 K. So, 3 cage is unstable at room temperature and is barely suitable for practical applications.
We also stress the fact that the kinetic stability of any cage compound is determined by the energy barrier preventing its decomposition rather than the strain energy enclosed in its framework. For example, methylcubanes demonstrate an inverse relationship between their strain energies and kinetic stabilities. Moreover, initial processes leading to the CL-20 decomposition do not necessarily involve any framework transformation. For these reasons, cage strain energy is not a suitable measure of the stability of the CL-20 derivatives. Although the CL-20 derivatives containing silicon possess higher strain energies, they are not necessarily kinetically unstable.
Within the framework of this project, we consider pyrolysis mechanisms, stability, and reactivity of two promising silicon CL-20 derivatives as well as their dimers. We obtain that the presence of silicon atoms in the cage changes the mechanisms of initial pyrolysis step, but does not significantly reduce the stability of the cage. In addition, Si-containing cages are more prone to dimerization. On the other hand, the simultaneous presence of silicon and fluorine atoms results in the compound instability. With regard to the presented results, the 2 compound seems to be the most attractive structure. It demonstrates higher crystalline densities, decomposition reaction heats, detonation velocities, detonation pressures, and explosion temperatures than the pristine CL-20. At the same time, according to the presented results, its kinetic stability, frontier orbitals, and chemical reactivity are very similar to the CL-20 characteristics.
Various nitrogen crystals
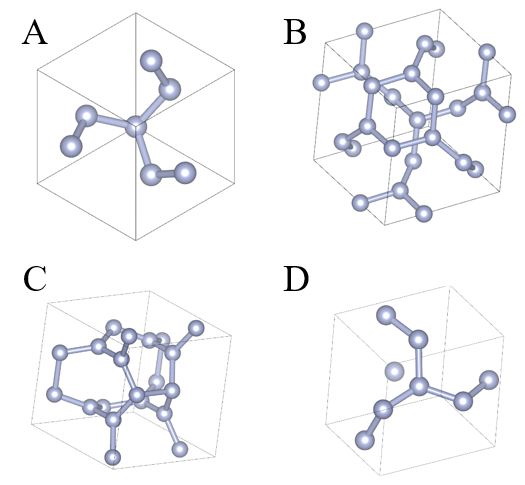
Within the framework of this project, several nitrogen crystalline phases with the following symmetries I213, I-43m, Pba2, and P212121 were examined. The unit cells of these crystals are shown in Figure 12.
|
|
Figure 12 – Unit cells of various nitrogen crystalline phases: I213 (A), I-43m (B), Pba2 (C), and P212121 (D). |
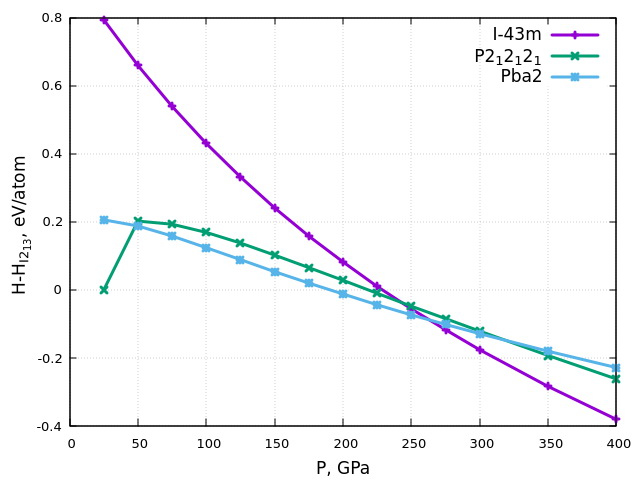
Figure 13 shows the pressure dependence of enthalpy per atom of a unit cell of the nitrogen crystalline phases considered in the range of 25–400 GPa. For clarity, in this Figure, the enthalpy of the I213 phase is taken as zero for every pressure value.
|
|
Figure 13 – Dependence of the enthalpy of various crystalline nitrogen phases, measured from the enthalpy of phase I213, on pressure. |
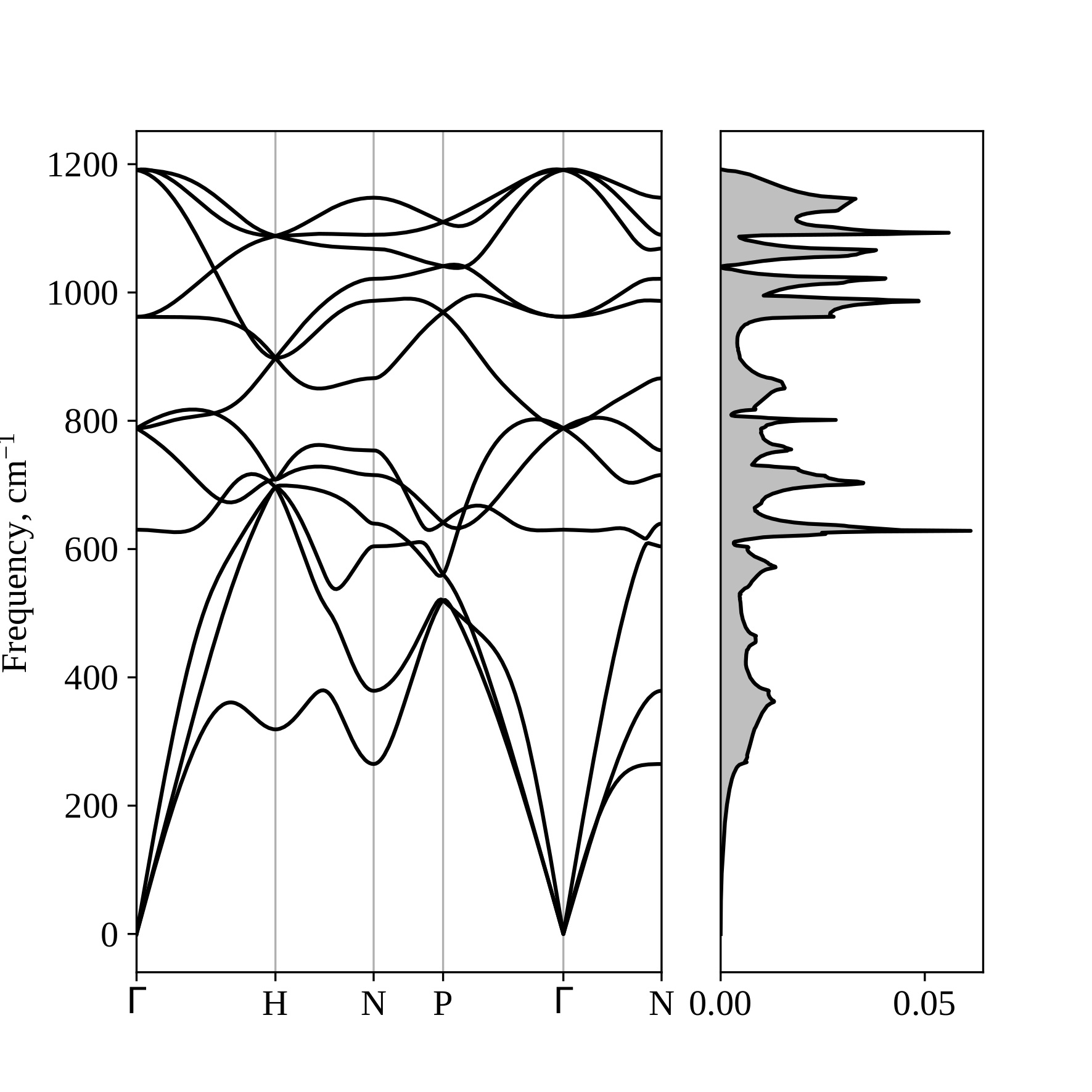
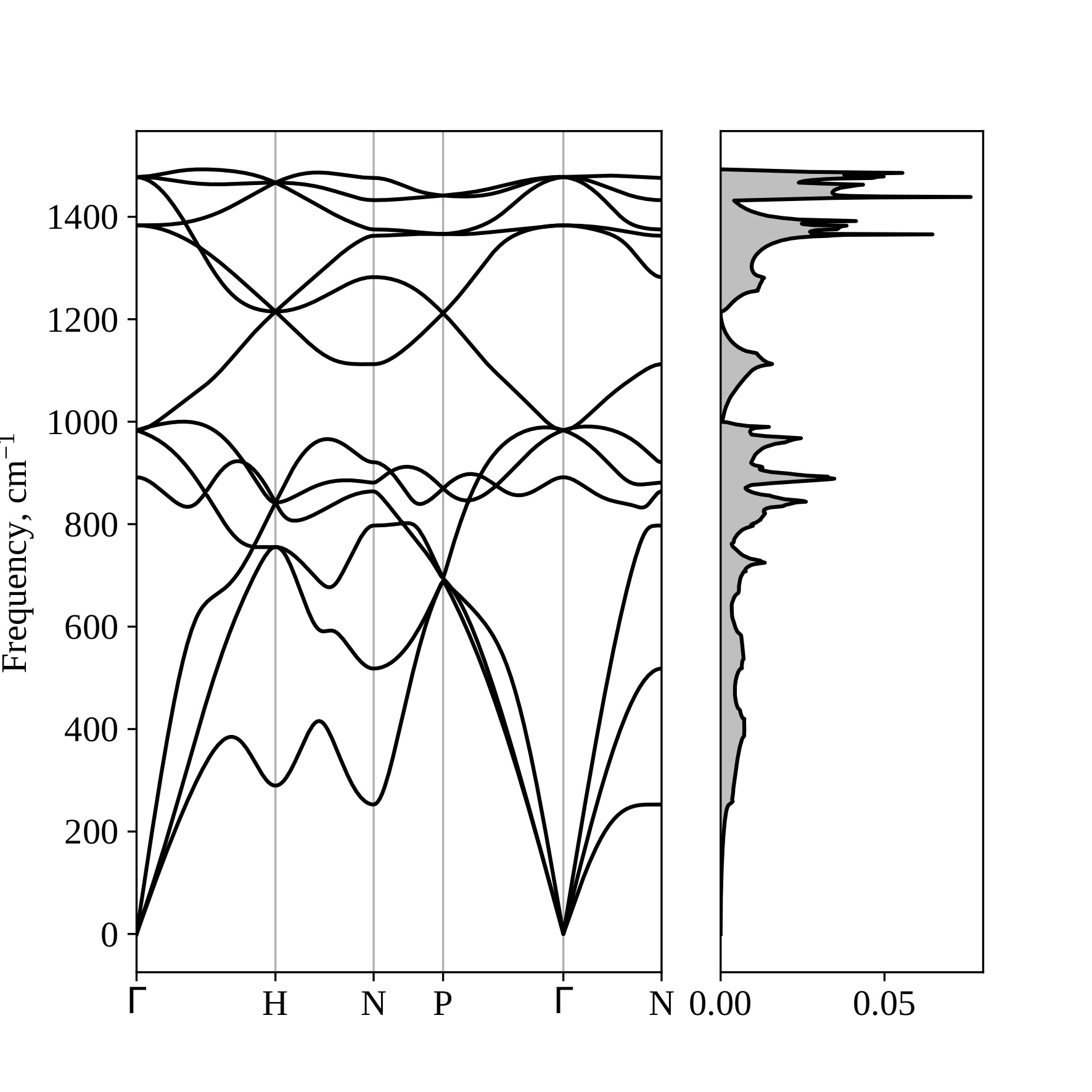
It can be seen that the nitrogen phase with I213 symmetry is energetically preferable among the nonmolecular nitrogen phases at pressures lower than ≈175 GPa. At pressure equals 25 GPa, the phase P212121 transforms to I213; therefore, the enthalpy values at this pressure coincide for these phases. In the range from ≈175-250 GPa, the phase with Pba2 symmetry is energetically favorable, and at higher pressures, the I-43m phase is preferable. Phase I213 is dynamically stable in the pressure range considered, as evidenced by the absence of imaginary frequencies in its phonon spectra (see Figure 14 at P = 25 GPa, Figure 15 at P = 175 GPa).
|
|
Figure 14 – Phonon spectrum and phonon density of states for the crystalline phase of nitrogen with I213 symmetry at a pressure of 25 GPa. |
|
|
Figure 15 – Phonon spectrum and phonon density of states for the crystalline phase of nitrogen with I213 symmetry at a pressure of 175 GPa. |
Using the VC-NEB (variable cell NEB) technique, a transition barrier of the nitrogen phase with I213 symmetry to the molecular phase was determined at a pressure of 50 GPa. The value of this barrier turns out to be quite high and equals to 1.2 eV, which indicates the possibility of experimental synthesis of this metastable crystalline form of nitrogen.
Topological rules and stability of nitrogen closed cages
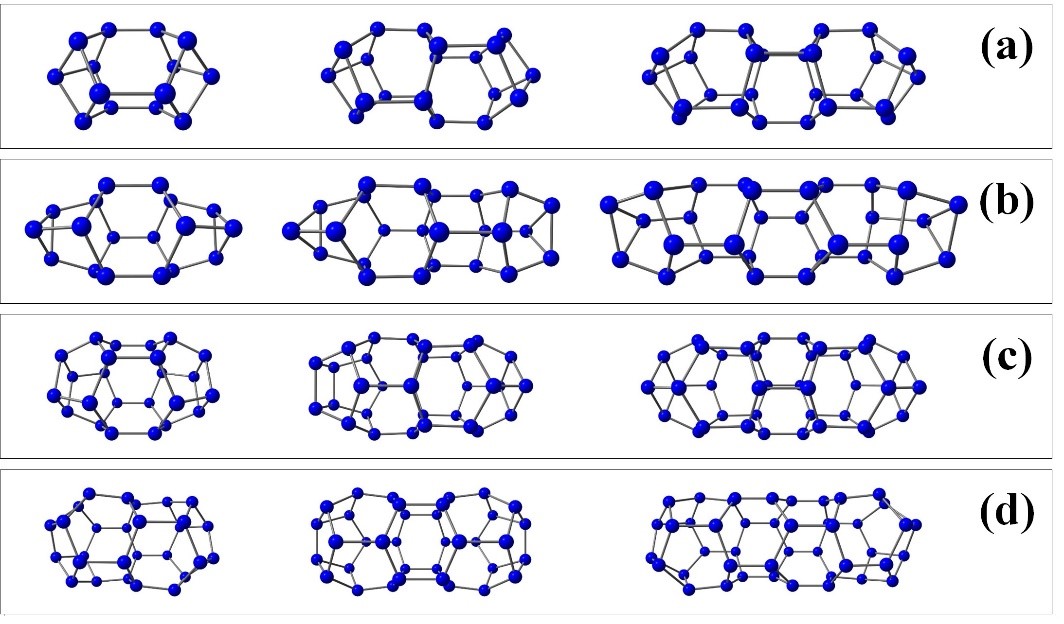
We carried out a systematic study of the structure and stability of Nn fullerene-like cages. Our goal was to identify the general relationships between topology and stability of nitrogen buckyballs. Unlike previous calculations, we focused on the pyrolysis mechanisms and the corresponding activation barriers. We identified the most stable Nn cages and explained the reasons for their stability. We constructed nitrogen analogs of low-energy isomers of carbon fullerenes from C20 to C120. All possible isomers C20 – C32, as well as all low-energy isomers C34 – C120, were used as the initial sample geometry for all-nitrogen cages. Note that the typical length of a single N–N bond (1.45 Å) is close to C–C bonds’ lengths in the fullerenes under consideration. However, all the corresponding Nn systems lose their fullerene-like shape when the geometry is optimized. The only exceptions were the clusters N20 and N24, which did not contain adjacent hexagons. For all other cages, optimization leads to breaking those N–N bonds that are common for the neighboring hexagons and the nitrogen cage’s subsequent destruction. Thus, we conclude that the presence of adjacent hexagons leads to cage instability. The only known Nn cages with the adjacent hexagons are tubular structures. The structure of the first members belonging to Nn tubular systems is shown in Figure 16. The extremely small diameters of these tubes provide stability due to the curvature effects and concave hexagon shapes. Note that the large diameter Nn tubular structures are unstable.
|
|
Figure 16 – Examples of tubular nitrogen clusters with adjacent hexagons. Nitrogen nanotubes with wall surface formed by three hexagons that contain caps at the ends closed by one (a) and three (b) nitrogen atoms. Nitrogen nanotubes with wall surface formed by four hexagons that contain caps at the ends closed by four (c) and two (d) nitrogen atoms. |
Many hexagons on the fullerene surface lead to a small curvature and large size of the system. To avoid adjacent pentagons, carbon clusters Cn should be sufficiently large (n ≥ 60). In contrast, the hexagons on the surface of nitrogen clusters should be separated by pentagons or smaller polygons, and therefore the fracture of the hexagons should be small. Thus, stable nitrogen clusters Nn should have a significant curvature and small size (n ≤ 24). Inspired by small carbon fullerenes and fullerene-like cages, we have constructed the corresponding nitrogen structures. All the nitrogen cages satisfying the isolated hexagon rule that are true local energy minima are presented in Figure 17.
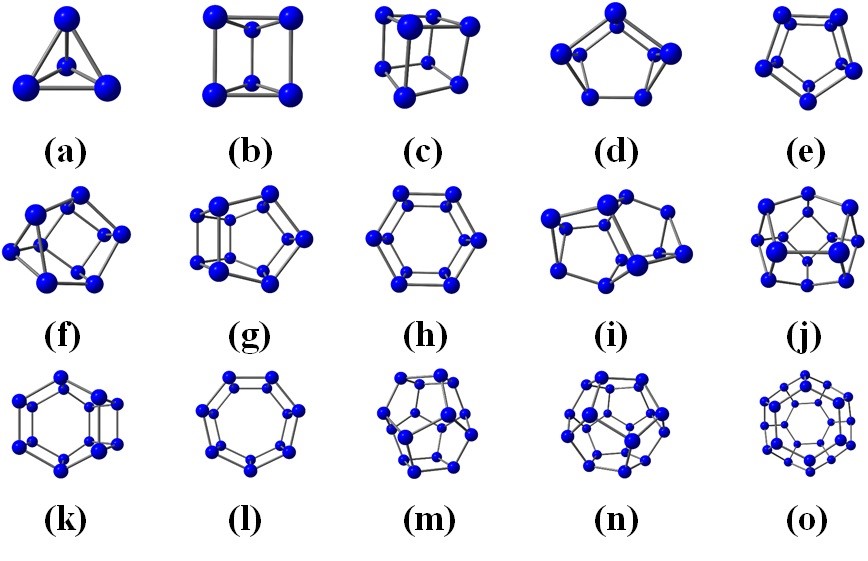
|
|
Figure 17 – Nitrogen cage structures with isolated hexagons. Atomic structures (a)-(e) are stable, while other systems possess low stability. |
These systems’ kinetic stability was investigated using ab initio molecular dynamics (AIMD) at T = 700 K for one picosecond. Only the five smallest nitrogen clusters, labeled as (a) - (e) in Figure 17, conserved their identity during the AIMD simulation. More massive clusters are decomposed. Therefore, they should be considered as low-stable systems. Note that N–N bonds in all truly stable and low-stable cages are longer than 1.4 Å, indicating that the bond type is single. To get a complete understanding of the origin of the stability of nitrogen cages, we analyzed the mechanisms of their pyrolysis. We adopted the reaction mechanism from the ab initio molecular dynamics. Knowing the reaction mechanism, we perform an intrinsic reaction coordinates calculation. The preliminary transient geometries obtained from the AIMD simulations are further optimized to find the valid transient configurations. One can define two distinct groups of systems: “stable” with energy barriers more than 0.5 eV and “low-stable” characterized by the value of energy barriers less than 0.25 eV.
Pyrolysis of five “stable” nitrogen cages is analyzed in more detail. Rate-dependent reaction steps, the corresponding transition states, and products are shown in Figure 18.
.jpg)
|
.jpg)
|
.jpg)
|
.jpg)
|
.jpg)
|
|
|
Figure 18 – Pyrolysis mechanisms of stable nitrogen clusters N4 (a), N6 (b), N8 (c), N8 (d), and N10 (e). |
|
Thermodynamic descriptors (activation enthalpies and Gibbs activation energies) at various temperatures are collected in Table 2. These descriptors weakly depend on temperature for all clusters since the initial and transition configurations are not so different, and their vibrational energies and entropies slightly differ from each other.
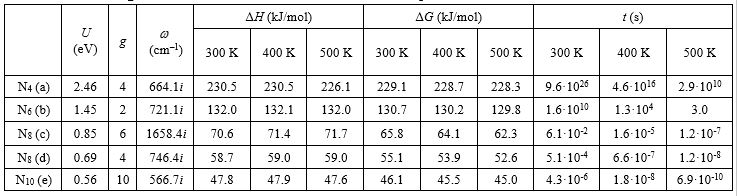
|
|
Table 2 – Characteristics of the decay of “stable” nitrogen cages Nn: energy barriers U, degeneracy factors g, imaginary frequencies of the transition state ω, activation enthalpies ΔH, activation Gibbs energies ΔG, and mean lifetimes t at different temperatures. |
According to Table 2, only N4 and N6 clusters have a reasonable lifetime at room temperature. Thus, they should be considered as the most promising high-energy-density structures. More extensive systems need additional stabilization with low temperature or high pressure.
Nitrogen astralene molecules
The diameter of the nanotube determines the shape of the cap that closes it. Four types of caps are known, consisting of one, two, three, or four nitrogen atoms. Based on these nanotubes, three types of astralenes were constructed, shown in Figure 19.
.jpg)
|
| (a) |
.jpg)
|
| (b) |
.jpg)
|
| (c) |
|
Figure 19 – Examples of nitrogen astralenes with tetrahedral (a), cubic (b), or hexagonal (c) core. |
Each astralene consists of four, five, or six intergrown halves of nanotubes with a common center so that a common core is formed in the central part of the system. Depending on the nanotubes’ diameter, it has a tetrahedral, cubic, or hexagonal shape, making it possible to classify astralenes into tetrahedral, cubic, and hexagonal, respectively. Each type of astralene can include certain types of caps at the ends of nanotubes: tetrahedral (one or three atoms), cubic (two or four atoms), hexagonal (from one to four atoms). The nanotube section between the core and the cap can include m hexagonal fragments (m = 0, 1, 2, etc.). An increase in m leads to a decrease in the stability of the system. The limiting value of m depends on the core shape and the diameter of the nanotubes. We restrict ourselves to the case of regular astralenes, characterized by the same m value and the same type of closing cap for each cylindrical branch of the nitrogen system. Thus, astralene can be denoted as XL(m,k), where X = T (tetrahedral), C (cubic) or H (hexagonal) describes the shape of the atomic core, L is the number of atoms in the core, m represents the length of cylindrical fragments, and k = 1,2,3 or 4 denotes the type of the cap, consisting of k nitrogen atoms. Hexagonal astralene includes intergrown nanotubes of different diameters, making it possible to use all existing types of closure caps. Several isomers differ in the orientation of the closing caps consisting of two atoms for the cubic astralenes. Such isomers will be denoted by CL(m,k)-r, where r = 0 ÷ 3 is the number of caps rotated 90 degrees relative to the “correct” position (see Figure 20).
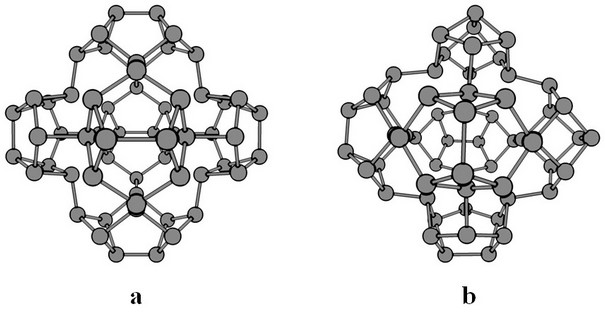
|
|
Figure 20 – Various isomers of cubic astralenes: C48(0,2): C48(0,2)-0 (a) and C48(0,2)-3 (b). |
The astralene atomic structure was found as a result of the geometric optimization of these systems. The average nitrogen-nitrogen bond length is in the range 1.404 - 1.553 Å, which is significantly higher than double N–N bonds with a length of 1.25 Å and is comparable with a single N–N bond length of 1.45 Å. Astralene cores are characterized by shorter bond lengths than the peripheral fragments. We calculated the binding energies of nitrogen astralenes. The tetrahedral astralene T24(2,3) (Eb = 3.12 eV/atom) has the highest binding energy, and T24(0,1) (Eb = 2.59 eV/atom) has the lowest binding energy. The “rotated” arrangement of one cap for cubic astralene C48(0,2)-1 lowers the binding energy, while the presence of four “rotated” caps insignificantly increases the stability of the entire structure. Similar behavior is observed for the nitrogen structures C56(0,2)-r. The calculation of the HOMO-LUMO gap showed that cubic astralene C48(0,2)-0 (5.05 eV) has the highest value, and T24(0,1) (2.59 eV) has the lowest value. The nitrogen system C56(0,4) is also distinguished, with the HOMO-LUMO gap of 4.82 eV that is quite large for cubic astralenes. As m increases, the gap decreases to 3.73 eV due to the incorporation of additional hexahedral fragments. The chemical reactivity analysis showed that the C48(0,2)-0 system possesses the highest chemical hardness. Note that the electrophilicity index C48(0,2)-0 is higher than the corresponding value of a similar astralene core with another type of closing cap C48(0,4). It should be noted that the astralene C56(0,2)-1 has the highest electrophilicity index and is the most chemically soft compound among the other cubic astralenes C56(0,2)-r. However, it possesses a lower electronegativity compared to astralene C56(0,2)-0. For cubic astralenes CL(m,4), an increase in the electrophilicity index is observed with an increase in the number of atoms in the core L. No monotonic dependence of the electronic characteristics and chemical reactivity of astralene on its molecular weight is observed.
Nitrogen astralene covalent crystals
New allotropic forms of non-molecular nitrogen based on astralenes were constructed. The elementary cell of the simplest crystalline astralene contains only the molecular astralene core. The supercells of more complex astralene crystals also contain cylindrical fragments, connecting with neighboring cells’ processes and forming a connection between the cores. We have limited ourselves to the case of the simplest astralene. We choose the cubic astralene core as the elementary cell. In this case, calculations showed that it is possible to obtain crystals with the space group Pm-3m containing 48, 56, and 64 atoms (Figure 21).
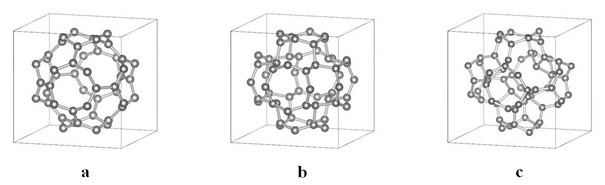
|
|
Figure 21 – Unit cells of cubic nitrogen astralenes: AC48 (a), AC56 (b), AC64 (c). |
For tetrahedral astralenes, the use of only core is not enough to form a stable system. However, the crystal system, in which cylindrical fragments are embedded between the cores, turned out to be stable. Thus, the first stable diamond-like crystalline nitrogen system with the space group Fd-3m, containing 144 atoms per unit cell, was obtained. The electronic characteristics of nitrogen crystal structures are presented in Figure 22.
.jpg)
|
.jpg)
|
.jpg)
|
|
Figure 22 – Band structure and the density of electronic states of nitrogen astralene crystals: AC48 (a), AC56 (b), and AC64 (c). |
Carbon nanotubes for stabilization of nitrogen nanostructures
We placed inside carbon zigzag and armchair nanotubes various unstable nitrogen systems under ambient conditions, such as nitrogen zigzag and armchair nanotubes, as well as initially unstable nitrogen fullerenes N36 and N60. Examples of considered structures are shown in Figure 23.
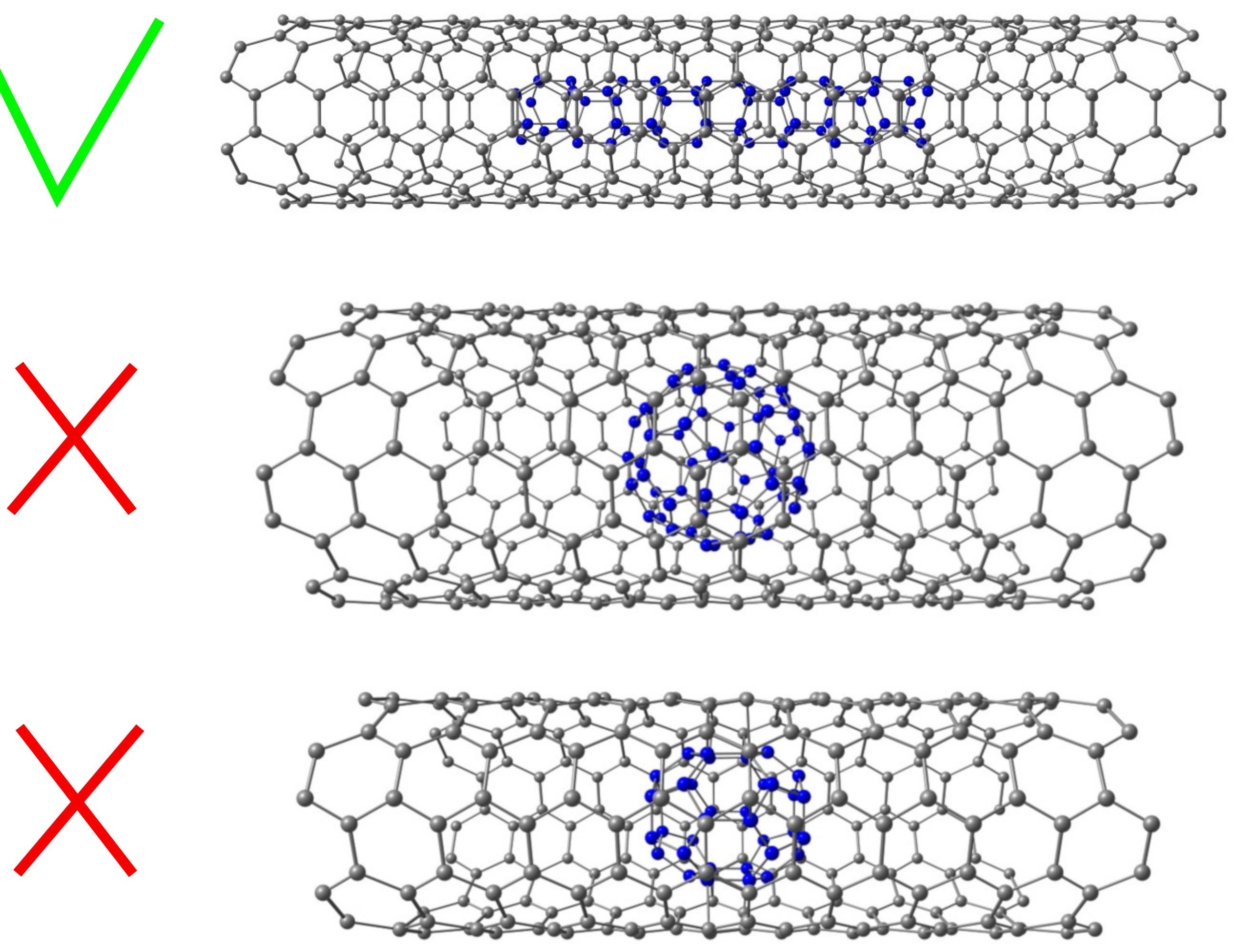
|
|
Figure 23 – The model geometries of simulated structures. Top to bottom: nitrogen nanotube inside carbon tube; N60 fullerene inside the carbon tube; N36 fullerene inside the carbon tube. |
Our calculations showed that only zigzag nitrogen nanotubes (3,0) and (4,0) were thermodynamically stable among the considered nitrogen systems encapsulated inside the carbon nanotubes. We found that the binding energy varies in the range of 3.5 - 4 eV/atom, depending on the diameters of the outer carbon and inner nitrogen nanotubes. Among all considered nitrogen systems encapsulated inside the carbon nanotube, the most energetically feasible is (3,0) zigzag nitrogen nanotube. The values of the bond lengths linter and lintra, corresponding to the interlayer and intralayer N-N bonds, obtained as a result of the geometrical optimization are linter = 1.488 Å, lintra = 1.46 ± 0.03 Å for nanotubes of type (3,0), and linter = 1.472 Å, lintra = 1.44 ± 0.02 Å for nanotubes of type (4,0). These values are close to the experimental single N-N bond length of 1.45 Å. The electronic properties of the structures under consideration are mainly determined by the external carbon nanotube’s electronic properties. When a carbon nanotube is a semiconductor, the introduction of a nitrogen nanotube inside it can lead to the appearance of allowed energy levels inside the bandgap, which is similar to functional doping. Finally, to estimate the stored energy in our stable nitrogen nanotubes, we determine the reaction energy ΔE[NNT→n·N2] ≈ 2 eV/atom. We found that carbon nanotubes can stabilize some nitrogen systems that are unstable under ambient conditions. It is shown that the zigzag nitrogen nanotubes of small diameter placed inside the carbon nanotubes become thermodynamically stable under ambient conditions. The numerical simulation confirmed that such systems could release a large amount of energy during the decomposition into isolated nitrogen molecules. We believe that the obtained results provide one more step toward nitrogen high-energy-density materials.
Functional doping of cubic gauche nitrogen surface
The surface of the cubic gauche nitrogen (cg-N) contains atoms with edged free bonds, which makes the structure unstable. Therefore, it is of particular interest to investigate the surface stabilization by filling dangling bonds by various radicals. We consider a surface in which nitrogen atoms located in the upper layer with dangling bonds are doped with CH3 radicals (see Figure 24).
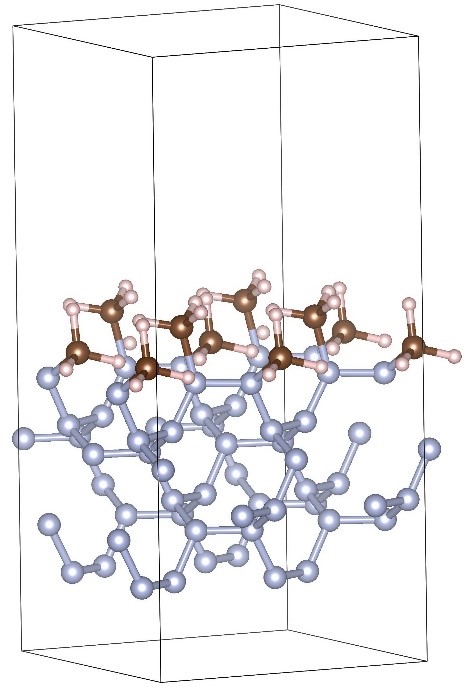
|
|
Figure 24 – Functional doping of cubic gauche nitrogen surface by CH3 functional groups. |
The surface was modeled by a structure periodic along the X and Y-axes with the size of 2×2 (in unit cells) along these axes. On the Z-axis, the system is not periodic. This is achieved by using a sufficiently large area of vacuum space along this axis. The distance between the nearest atoms in two adjacent unit cells along the Z-axis does not exceed 9.5 Å. Along the Z-axis, the structure consists of six nitrogen layers (approximately 1.5 unit cells of the cubic gauche nitrogen in the z-direction). The atoms of the three lower layers are fixed. The nitrogen atoms with free bonds located in the upper layer are bound with CH3 radicals. The calculation results show that such a configuration of the surface of the cubic gauche nitrogen is stable. Thus, eliminating free bonds at the cg-N boundaries by the CH3 radicals helps to stabilize the structure. Calculations of the electron density of states show that such incorporation of CH3 radicals along the cg-N interface leads to the appearance of a region with allowed states in the bandgap. Such behavior is similar to the introduction of impurities into a semiconductor (Figure 25).
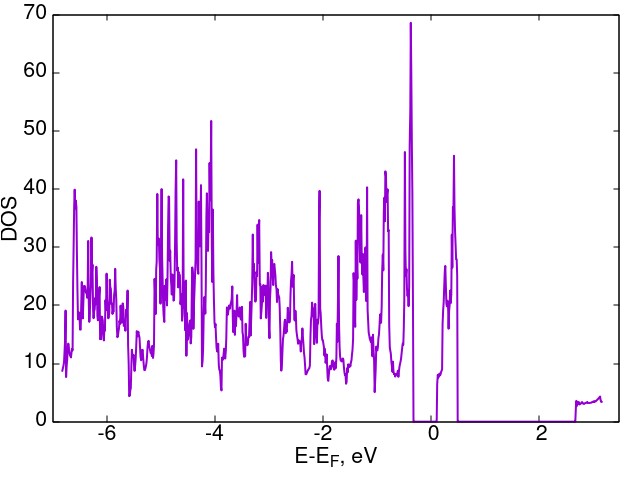
|
|
Figure 25 – Density of electronic states of CH3-doped cubic gauche nitrogen surface. |
Publications
For detailed information, see the following publications:
1. Katin K.P., Javan M.B., Kochaev A.I., Soltani A., Maslov M.M. Kinetic Stability and Reactivity of Silicon and Fluorine-Containing CL-20 Derivatives // ChemistrySelect. 2019. V. 4. P. 9659-9665. DOI: 10.1002/slct.201902583
2. Grishakov K.S., Katin K.P., Gimaldinova M.A., Maslov M.M. Stability and energy characteristics of extended nitrogen nanotubes: Density functional theory study // Letters on Materials. 2019. V. 9. P. 366-369. DOI: 10.22226/2410-3535-2019-3-366-369
3. Gimaldinova M.A., Zemenkov L.I., Merinov V.B. Stabilization of small nitrogen clusters via spatial constraint // Journal of Physics: Conference Series. 2020. V. 1435. P. 012062. DOI: 10.1088/1742-6596/1435/1/012062
4. Salem M.A., Katin K.P., Kaya S., Kochaev A.I., Maslov M.M. Interaction of dopants and functional groups adsorbed on the carbon fullerenes: Computational study // Physica E: Low-dimensional Systems and Nanostructures. 2020. V. 124. P. 114319. DOI: 10.1016/j.physe.2020.114319
5. Gimaldinova M.A., Katin K.P., Grishakov K.S., Maslov M.M. Kinetic stability of nitrogen cubane inside the fullerene cage: Molecular dynamics study // Fullerenes, Nanotubes and Carbon Nanostructures. 2020. V. 28. P. 304-308. DOI: 10.1080/1536383X.2019.1708730
6. Katin K.P., Grishakov K.S., Podlivaev A.I., Maslov M.M. Molecular hyperdynamics coupled with the nonorthogonal tight-binding approach: Implementation and validation // Journal of Chemical Theory and Computation. 2020. V. 16. P. 2065-2070. DOI: 10.1021/acs.jctc.9b01229
Acknowledgments
The presented study was performed with the financial support of the Russian Foundation for Basic Research (RFBR) (Grant No. 18-32-20139 mol_a_ved). We are grateful to DSEPY-RI for the organizational support and computational resources provision.